Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
Metabolismul proteinelor

2
Obiectivele Nevoile de proteină în alimentaţie. Starea dinamică a proteinelor. Valoarea biologică a proteinelor. Bilanţul azotat. Digestia proteinelor în stomac şi intestin. Endo- şi exopeptidazele, specificitatea de acţiune a proteinkinazelor. Proenzimele proteinkinazelor şi mecanismul convertirii lor la enzime active. Reglarea secreţiei sucului gastric, pancreatic şi intestinal. Absorbţia aminoacizilor în intestin. Putrefacţia aminoacizilor în intestin. Alimentaţia proteică parenterală. Compoziţia sucului gastric şi modificările lui în patologie

3
Proteinele – substanţe macromoleculare de natură polipeptidică.
Au un rol fundamental atît prin funcţiile lor structurale (matricea tuturor ţesuturilor) cît şi dinamice (rol de transport, control metabolic, medierea unor reacţii biochimice etc.)
 cît şi dinamice (rol de transport, control metabolic, medierea unor reacţii biochimice etc.)")
4
Necesarul de proteină în alimentaţie
Sunt substanţe nutritive deosebit de importante: sunt singura sursă de N asimiabil de organism; sunt furnizatoare de AA esenţiali Aportul zilnic exogen de proteine este: La un adult – g La un efort fizic – g La copii – g

5
Starea dinamică a proteinelor
Proteinele din organism se reînnoiesc permanent. Pentru menţinerea constantă a proporţiei lor în ţesuturi, vitezele de sinteză şi de degradare a proteinelor trebuie să fie egale, ceea ce constituie o stare dinamică staţionară.

6
Vitezele de reînnoire a proteinelor se exprimă prin timpul de înjumătăţire (T1/2), ce diferă în diferite organe. De exemplu: T1/2 Pr musculare = 30 zile; T1/2 Pr hepatice = 5-6 zile; T1/2 enzimelor = ore, minute.

7
Bilanţul azotat- BA Starea dinamică a proteinelor este reflectată de BA. BA al organismului - raportul dintre cantitatea de N îngerat şi cantitatea de N excretat din organism (urină, fecale, salivă, gl.sudoripare) exprimat în g/24 ore. Deosebim 3 tipuri de BA: echilibrat - Nîng = Nexcr; pozitiv – cantitatea de N îngerat > N eliminat (specific pentru organisme în creştere, femeile în perioada de gestaţie, lactaţie); negativ – cantitatea de N ingerat< N eliminat. BA negativ se întîlneşte la persoanele de vârsta a treia şi în patologii: cancerul, însoţit de caşexie, tuberculoză, nefrite, combustii, înaniţie.
 exprimat în g/24 ore. Deosebim 3 tipuri de BA: echilibrat - Nîng = Nexcr; pozitiv – cantitatea de N îngerat > N eliminat (specific pentru organisme în creştere, femeile în perioada de gestaţie, lactaţie); negativ – cantitatea de N ingerat< N eliminat. BA negativ se întîlneşte la persoanele de vârsta a treia şi în patologii: cancerul, însoţit de caşexie, tuberculoză, nefrite, combustii, înaniţie.")
8
Starea funcţională normală a organismului depinde de aportul de proteine (AA) din exterior.
De menţionat, că ea este influenţată nu numai de cantitatea ei ci şi de calitatea proteinelor alimentare, ce au valoarea biologică diferită.

9
Valoarea biologică a proteinelor
VB înaltă posedă proteinele ce au o componenţă structurală mai apropiată de cea a proteinelor umane şi care pot fi hidrolizate complet în TGI. VB a proteinelor alimentare este determinată de 2 factori: AA ce întră în componenţa lor - de cantitatea AA indispensabili - AA care nu se sintetizează în celulele organismului ( 8 AA: Val, Leu, Ile; Liz, Met, Tre, Tri, Fen şi AA semidispensabili - Arg, His). 2. capacitatea organismului de a asimila AA proteinei date.
. 2. capacitatea organismului de a asimila AA proteinei date.")
10
Valoarea biologică a proteinelor
Lipsa sau carenţa unui AA indispensabil din alimente duce la afectarea absorbţiei celorlalţi AA. În aceste cazuri creşterea, dezvoltarea şi funcţionarea organismelor vii e determinată de acea substanţa indispensabilă, care e absorbită din alimente în cantitate cea mai mică (legitatea de minimum a lui Liebig).
.")
11
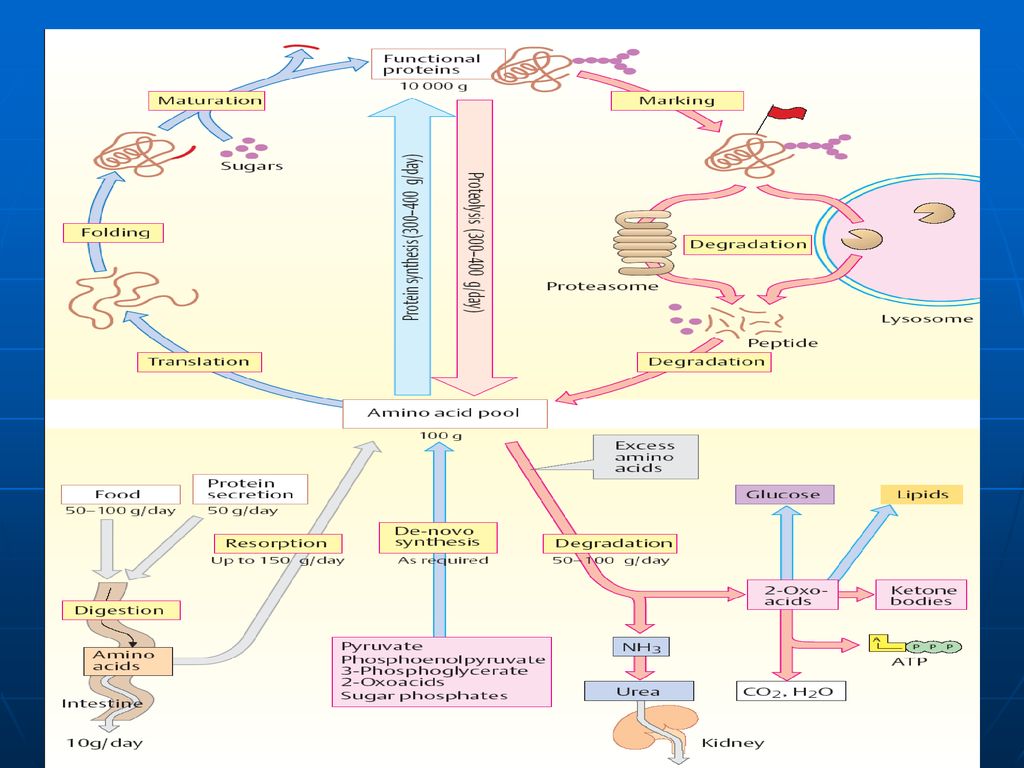
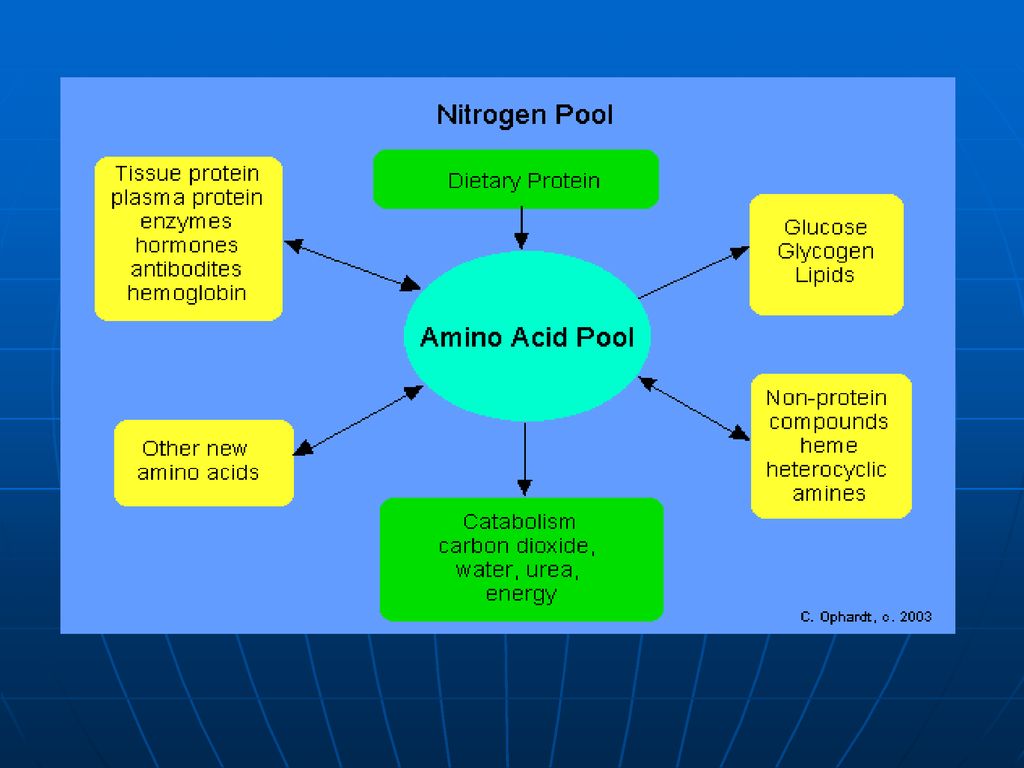
Fondul metabolic comun (FMC) al AA
Spre deosebire de glucide, ce se acumulează în muşchi şi ficat, sau lipide, ce se depun în ţesutul adipos, proteinele şi AA nu depozitează. Unica rezervă a lor o prezintă FMC al AA FMC- este totalitatea AA liberi în organism de origine atât exogenă (alimentele) cât şi endogenă (degradarea proteinelor) care sunt utilizaţi pentru sinteza proteinelor de către celule. Ei prezintă circa 30g din totalitatea de 15kg de proteinele ale organismului. Cantitatea majoră o constituie AA sanguini (0,35-0,65g/l). În condiţii extremale (nu este aport de AA din mediul ambiant) FMC este completat prin degradarea proteinelor plasmatice şi ale ficatului.
 cât şi endogenă (degradarea proteinelor) care sunt utilizaţi pentru sinteza proteinelor de către celule. Ei prezintă circa 30g din totalitatea de 15kg de proteinele ale organismului. Cantitatea majoră o constituie AA sanguini (0,35-0,65g/l). În condiţii extremale (nu este aport de AA din mediul ambiant) FMC este completat prin degradarea proteinelor plasmatice şi ale ficatului.")
14
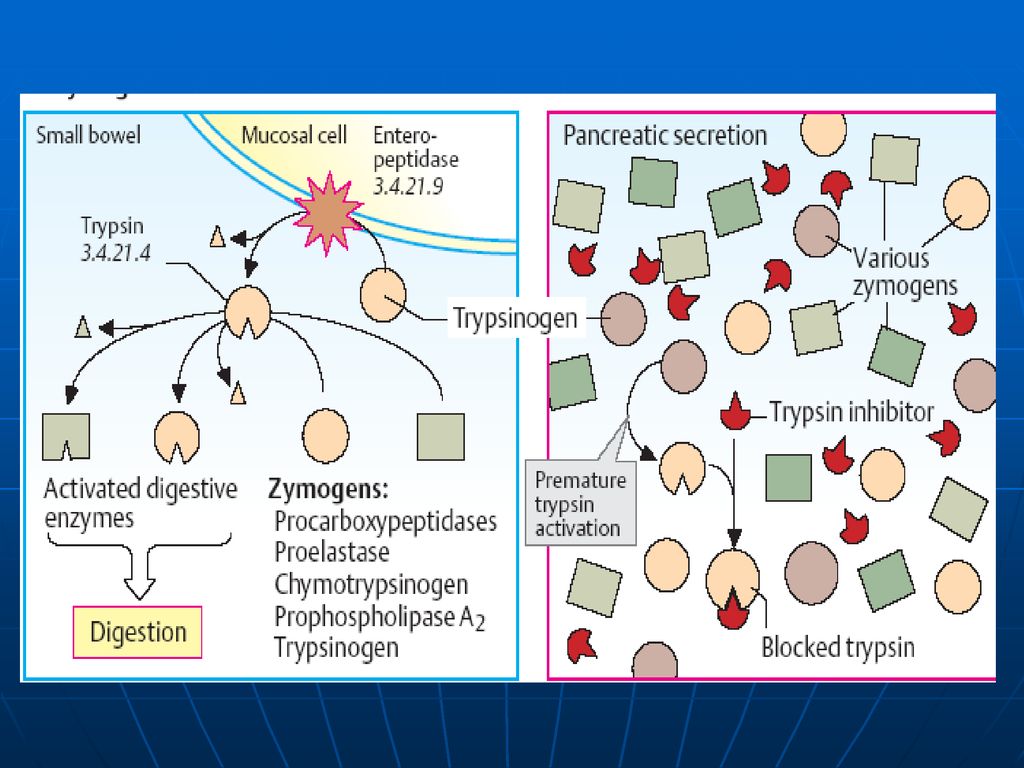
Digestia proteinelor în TGI
Digestia proteinelor are loc în stomac şi intestinul subţire sub acţiunea E proteolitice (hidrolaze) din sucul gastric, pancreatic şi intestinal. Toate aceste E catalizează hidroliza legăturii peptidice între ele există diferenţe de specificitate. sunt secretate de celulele producătoare în forme inactive – numite proenzime (zimogeni). Activarea are loc prin: proteoliză parţială (detaşarea unor oligopeptide de la capetele lor sau din interior, în urma căreia are loc formarea conformaţiei active a CA al E.) autocatalitic
 din sucul gastric, pancreatic şi intestinal. Toate aceste E catalizează hidroliza legăturii peptidice. între ele există diferenţe de specificitate. sunt secretate de celulele producătoare în forme inactive – numite proenzime (zimogeni). Activarea are loc prin: proteoliză parţială (detaşarea unor oligopeptide de la capetele lor sau din interior, în urma căreia are loc formarea conformaţiei active a CA al E.) autocatalitic.")
15
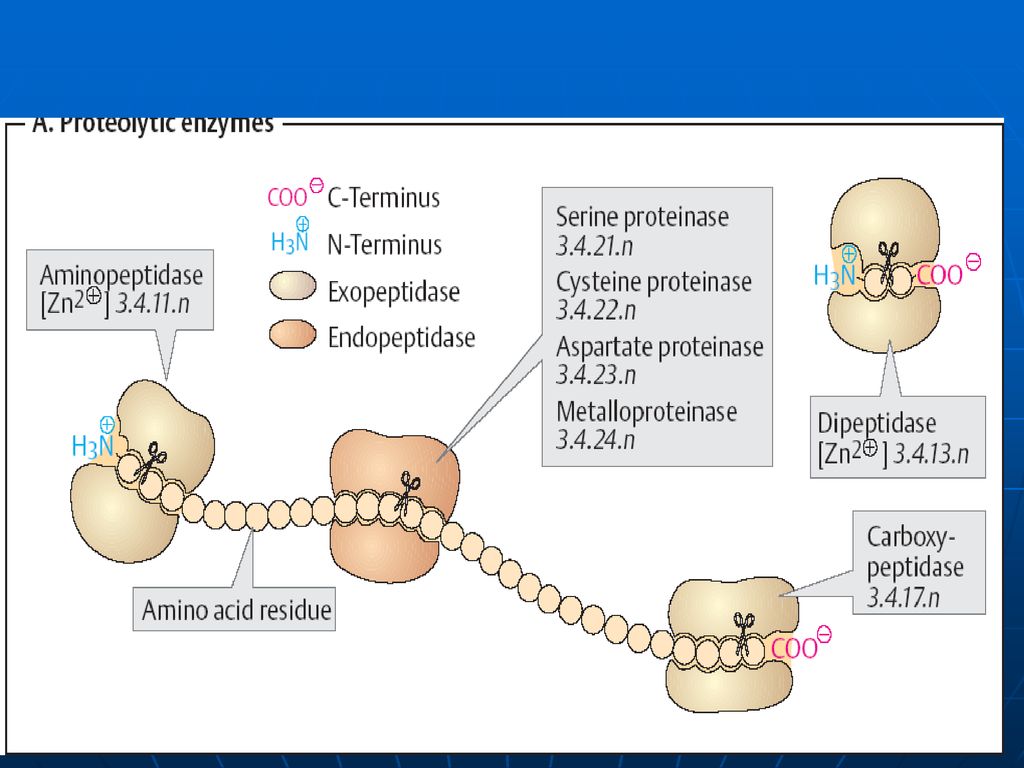
E proteolitice Se disting: endo- şi exopeptidaze.
Endopeptidazele – care asigură scindarea legăturilor peptidice din interiorul lanţurilor polipeptidice. Exopeptidazele – E ce scindează legăturile peptidice formate de AA terminali.

17
E proteolitice ale sucului gastric
Pepsina gastrixina renina (sugari).
.")
18
Pepsina se sintetizează de celulele principale ale mucoasei stomacului sub formă de pepsinogen. Pepsinogenul este activat la pepsina> 1. proteoliză parţială (H+ (HCl)) 2. autocatalitic H+ Pepsinogen → Pepsină -42 AA PH optim 1-1,5 este endopeptidază, Specificitatea - atacă legăturile peptidice la care participă – prin grupările aminice – AA aromatici şi într-o mică măsură - Met, Leu şi AA dicarboxilici
) 2. autocatalitic. H+ Pepsinogen → Pepsină. -42 AA. PH optim 1-1,5. este endopeptidază, Specificitatea - atacă legăturile peptidice la care participă – prin grupările aminice – AA aromatici şi într-o mică măsură - Met, Leu şi AA dicarboxilici.")
19
Gastrixina (pepsina C) – un analog structural al pepsinei.
pH-ul optim de acţiune ~3, deaceia activitatea ei predomină la copii. Specificitatea de acţiune se manifestă asupra legăturile peptidice din interiorul lanţurilor proteice, formate de a/a dicarboxilici. Chimozina (renina) este prezentă în sucul gastric al sugarilor. În prezenţa ionilor de Ca2+ chimozina transformă cazeina laptelui în paracazeină (hidrolizată apoi de pepsină). Punctul izoelectric al reninei – 4,5. În stomac, ca urmare a acţiunei hidrolitice specifice a pepsinei şi gastrixinei, din proteine se obţin polipeptide şi eventual oligopeptide, nu însă AA liberi.
 este prezentă în sucul gastric al sugarilor. În prezenţa ionilor de Ca2+ chimozina transformă cazeina laptelui în paracazeină (hidrolizată apoi de pepsină). Punctul izoelectric al reninei – 4,5. În stomac, ca urmare a acţiunei hidrolitice specifice a pepsinei şi gastrixinei, din proteine se obţin polipeptide şi eventual oligopeptide, nu însă AA liberi.")
20
Rolul HCl denaturarea parţială a proteinelor alimentare şi hidroliza proteinelor compuse; activarea pepsinogenului; menţinerea pH optim; acţiune antimicrobiană; participă la absorbţia Fe2+ Stimulează secreţia secretinei

21
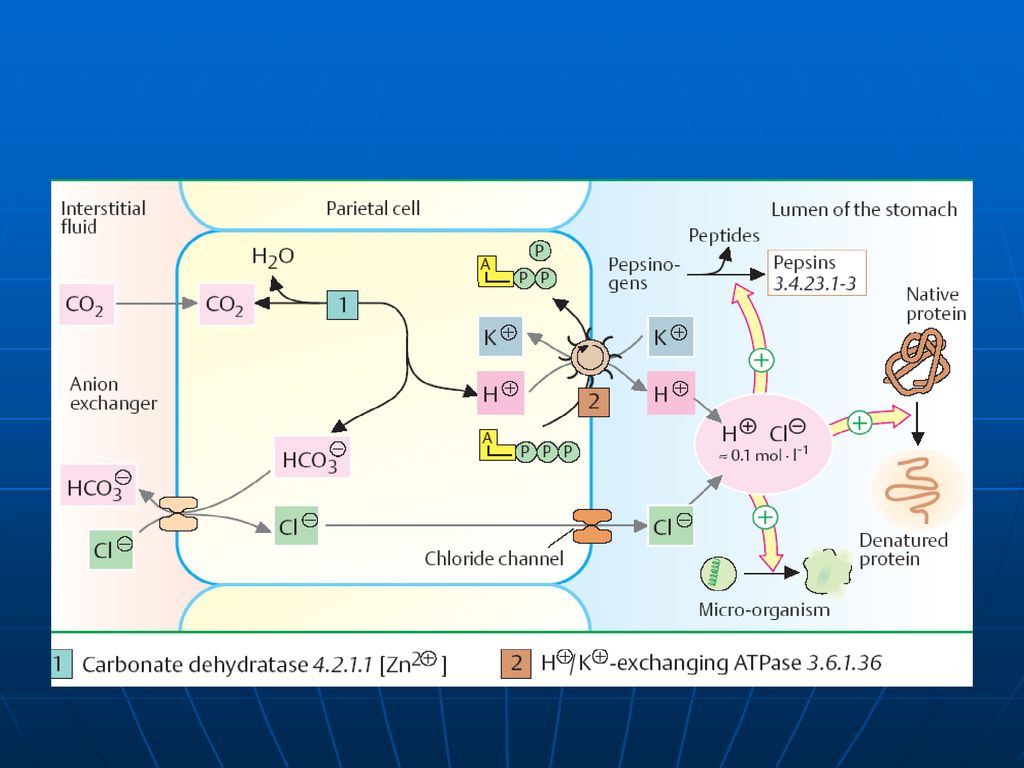
Sinteza HCl în stomac un proces complex asigurat de celulele secundare ale mucoasei. CA Sinteza acidului carbonic: CO2 + H2O ---→ H2CO3 disociază H2CO → HCO3ˉ + H Protonii H+ sunt transportaţi în lumenului stomacului printr-un mecanism asemănător cu transportarea protonilor dependent de ATP la funcţionarea ATP-azei din membrana internă a MC. Ionii de Clˉ provin din NaCl sanguin. NaCl +H2CO3 →NaHCO3 (în plasmă) +HCl (se secretează)
 +HCl (se secretează)")
22
Reglarea secreţiei HCl
În reglarea secreţiei HCl în mucoasa gastrică un rol important îi revine H- K- ATP-azei E e localizată în membrana apicală a celulelor epiteliale din mucoasă Este alcătuită din 2 subunităţi α (funcţie catalitică) şi o β subunitate (glicoproteid, ce determină localizarea E în membrană şi reglează funcţia ei de transport) Rolul: catalizează hidroliza ATP la ADP+P, cuplată cu sistemul de schimb al H intracelular pe K extracelulart
 şi o β subunitate (glicoproteid, ce determină localizarea E în membrană şi reglează funcţia ei de transport) Rolul: catalizează hidroliza ATP la ADP+P, cuplată cu sistemul de schimb al H intracelular pe K extracelulart.")
24
Secreţia HCl din celule este activat de:
Gastrină, histamină, acetilcholină Histamina acţionează nemijlocit, pe când gastrinele prin mărirea cantităţii de histamină activează adenilatciclaza, care la rîndul ei prin intermediul AMPc şi PK-aza activează carbanhidraza (e activă în formă fosforilată). În rezultat creşte cantitatea de H+ - ce se foloseşte la sinteza HCl. Secretina şi somatostatina inhibă secvenţa prin diminuarea formării gastrinelor (4 polipeptide sintetizate în partea pilorică a stomacului).
. În rezultat creşte cantitatea de H+ - ce se foloseşte la sinteza HCl. Secretina şi somatostatina inhibă secvenţa prin diminuarea formării gastrinelor (4 polipeptide sintetizate în partea pilorică a stomacului).")
25
Schema influenţei gastrinei la sinteza HCl

26
E proteolitice ale sucului pancreatic:
tripsina (endopeptidaza), chimotripsina (endopeptidaza), elastaza (endopeptidaza), carboxipeptidaza (exopeptidaza).
, chimotripsina (endopeptidaza), elastaza (endopeptidaza), carboxipeptidaza (exopeptidaza).")
27
Tripsina: Endopeptidază Se sintetizează sub formă de tripsinogen
este convertit în tripsină prin: 1.proteoliză limitată (îndepărtarea din capătul N-terminal a unui hexapeptid) sub acţiunea enterochinazei (E secretată de mucoasa intestinală) autocatalitic. Enterochinaza Tripsinogenul →Tripsina Ca2+ Specificitatea: hidrolizează legăturile peptidice cu participarea grupelor carboxil ale lizinei şi argininei. Tripsina participă şi la activarea altor E din lumenul intestinului.
 sub acţiunea enterochinazei (E secretată de mucoasa intestinală) autocatalitic. Enterochinaza. Tripsinogenul →Tripsina. Ca2+ Specificitatea: hidrolizează legăturile peptidice cu participarea grupelor carboxil ale lizinei şi argininei. Tripsina participă şi la activarea altor E din lumenul intestinului.")
29
Chimotripsina se sintetizează din chimotripsinogen:
sub acţiunea tripsinei (prin îndepărtarea a două dipeptide) autocatalitic. tripsinei Chimotripsinogen →chimotripsina Ca2+ Deosebim câteva forme de chimotripsine – α, δ şi p Specificitatea: hidrolizează legăturile peptidice formate de grupa –COOH a Phe, Tyr, Tri. scindează amide, esteri, derivaţi acil.
 autocatalitic. tripsinei. Chimotripsinogen →chimotripsina. Ca2+ Deosebim câteva forme de chimotripsine – α, δ şi p. Specificitatea: hidrolizează legăturile peptidice formate de grupa –COOH a Phe, Tyr, Tri. scindează amide, esteri, derivaţi acil.")
30
Elastaza Se obţine din proelastază (sub acţiunea tripsinei) Specificitatea: catalizează hidroliza legăturile peptidice formate de AA hidrofobi relativ mici: Gli, Ala, Ser. Carboxipeptidaza A: Este o exopeptidază Este o metaloproteină (E ce conţine Zn) Specificitatea: scindează legăturile peptidice formate de AA aromatici Atunci cînd ionul de Zn =>Ca – se declanşează activitatea esterazică Carboxipeptidaza B: - acţionează asupra legăturile peptidice din capătul C terminal, formate de Arg şi Lyz
 Specificitatea: scindează legăturile peptidice formate de AA aromatici. Atunci cînd ionul de Zn =>Ca – se declanşează activitatea esterazică. Carboxipeptidaza B: - acţionează asupra legăturile peptidice din capătul C terminal, formate de Arg şi Lyz.")
31
E sucului intestinal Aminopeptidazele: exopeptidaze Ala aminopeptidaza → specifică numai pentru Ala Leu aminopeptidaza → conţine Zn, pe care o poate activa Mn e specifică pentru toţi AA N terminali Dipeptidazele: glicil-glicină; prolinaza (COOH), prolidaza (NH). Sub acţiunea tuturor acestor enzime are loc scindarea totală a proteinei pînă la AA liberi.
, prolidaza (NH). Sub acţiunea tuturor acestor enzime are loc scindarea totală a proteinei pînă la AA liberi.")
32
Reglarea proteazelor Reglarea secreţiei enzimatice se face cu participarea următoarelor substanţe active: gastrina - stimulează secreţia pepsinogenului şi a HCl. Histamina - stimulează secreţie HCl.

33
sinteza şi secreţia sucului pancreatic e reglată de: secretină şi colecistokinină Secretina stimulează eliminarea unui suc pancreatic bogat în bicarbonaţi şi sărac în E, ce are menirea de a: neutraliza HCl, ce pătrunde cu bolul alimentar din stomac de a crea pH optim pentru acţionarea E pancreatice – 7,5-8,5. Sinteza secretinei în mucoasa duodenului este stimulată de HCl. Colecistokinina - stimulează eliminarea unui suc pancreatic bogat în E (stimulează contracţia vezicii biliare) şi sărac în bicarbonaţi.
 şi sărac în bicarbonaţi.")
34
Absorbţia are loc la nivelul intestinului subţire
este un proces activ cu solicitare de energie, cuplat cu transportul ionilor de Na. Absorbţia AA prin difuzie e limitată. Transportul în celulele epiteliale intestinale se efectuează cu ajutorul unor proteine specializate, numite translocaze. Există următoarele translocaze de grup: pentru AA neutri cu molecule mici pentru AA neutri cu molecule mari (a/a aromatici) pentru AA bazici şi cisteină pentru AA acizi pentru Pro şi hidroxiprolină După alimentaţie, concentraţia max de AA în sânge se înregistrează la o oră.
 pentru AA bazici şi cisteină. pentru AA acizi. pentru Pro şi hidroxiprolină. După alimentaţie, concentraţia max de AA în sânge se înregistrează la o oră.")
35
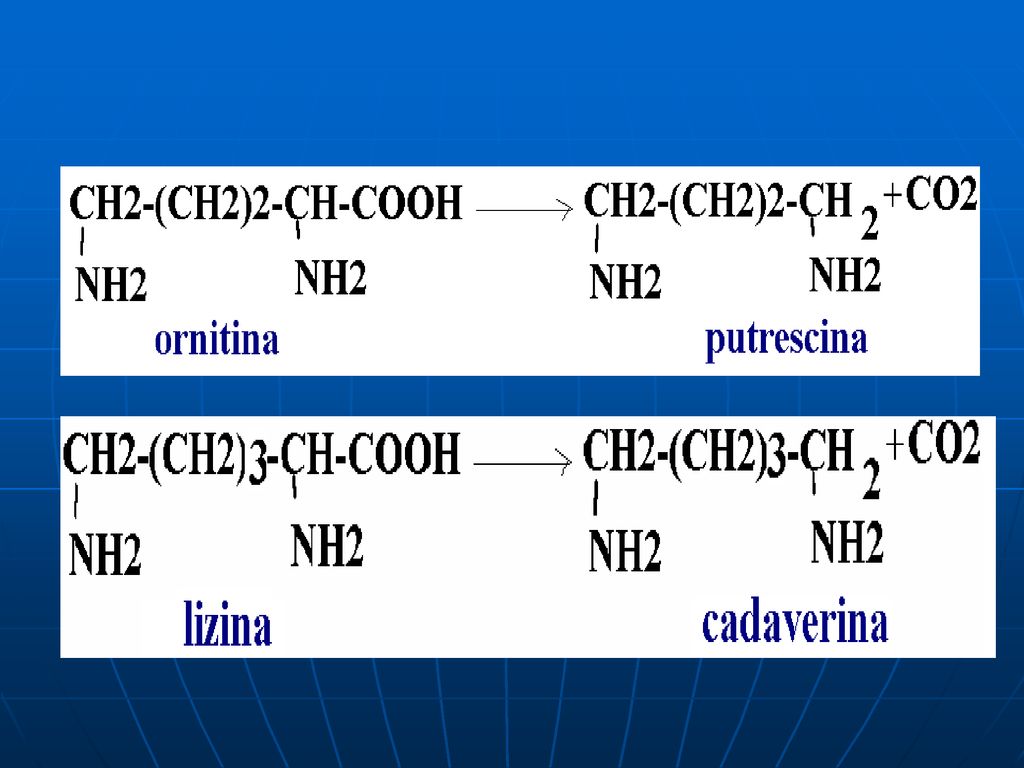
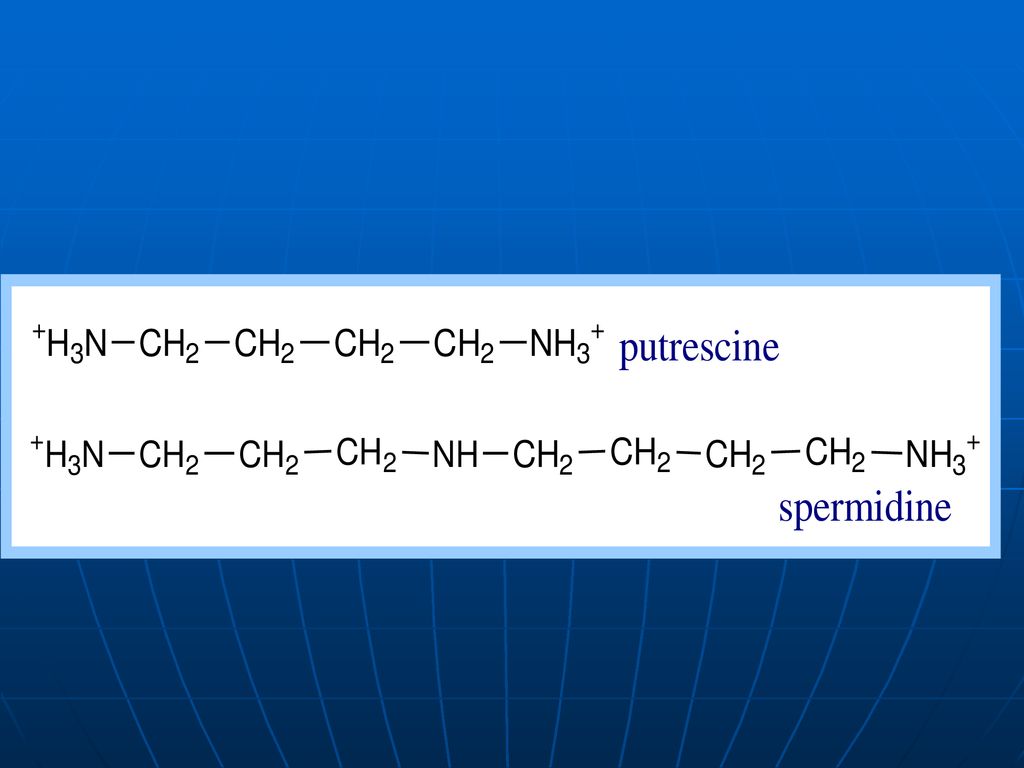
Putrefacţia AA în intestin
O parte din AA alimentelor este scindată de E microflorei intestinale, ce catalizează reacţii deosebite de cele din ţesuturi. Acest proces se numeşte putrefacţie. La scindarea Cis, Met (conţin sulf), în intestin se formează H2S, metilmercaptanul (CH3SH). Ornitina şi Lys se decarboxilează cu formarea aminelor - putrescina şi cadaverina.
, în intestin se formează H2S, metilmercaptanul (CH3SH). Ornitina şi Lys se decarboxilează cu formarea aminelor - putrescina şi cadaverina.")
38
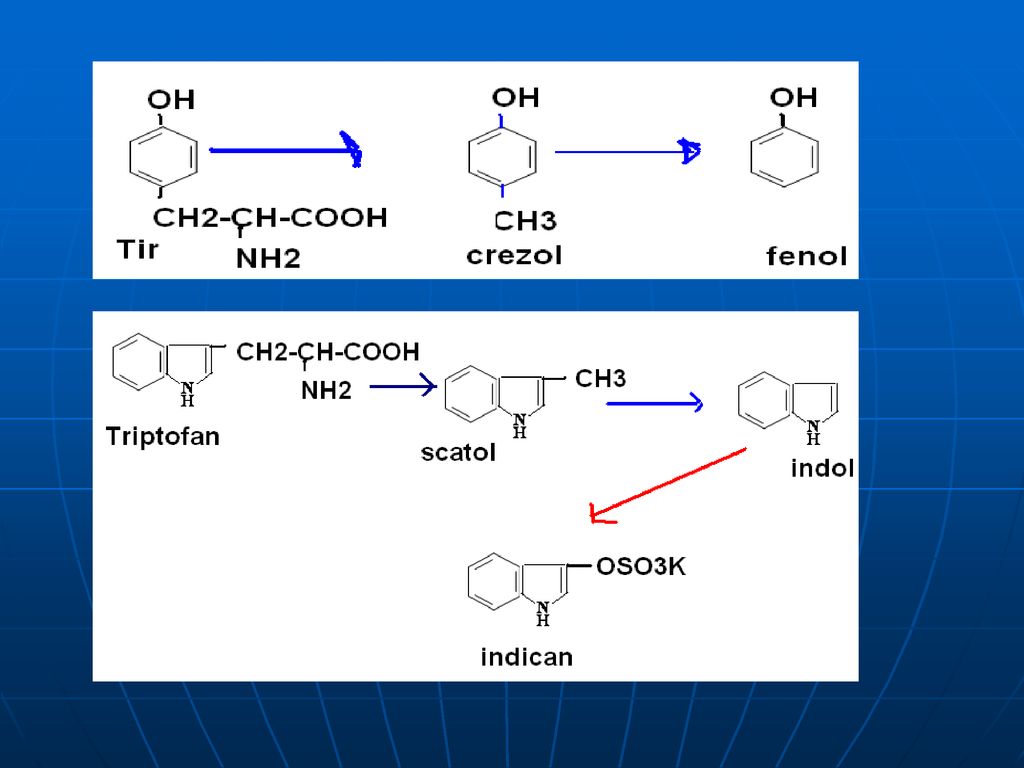
3. La o decarboxilare bacteriană din Phe, Tyr, Trp se formează aminele biogene corespunzătoare – feniletilamina, tiramina, triptamina. 4. Degradarea catenelor laterale ale AA ciclici duc la formarea produselor toxice: din Tyr se formează crezol, fenol; din Trn – scatol, indol.

40
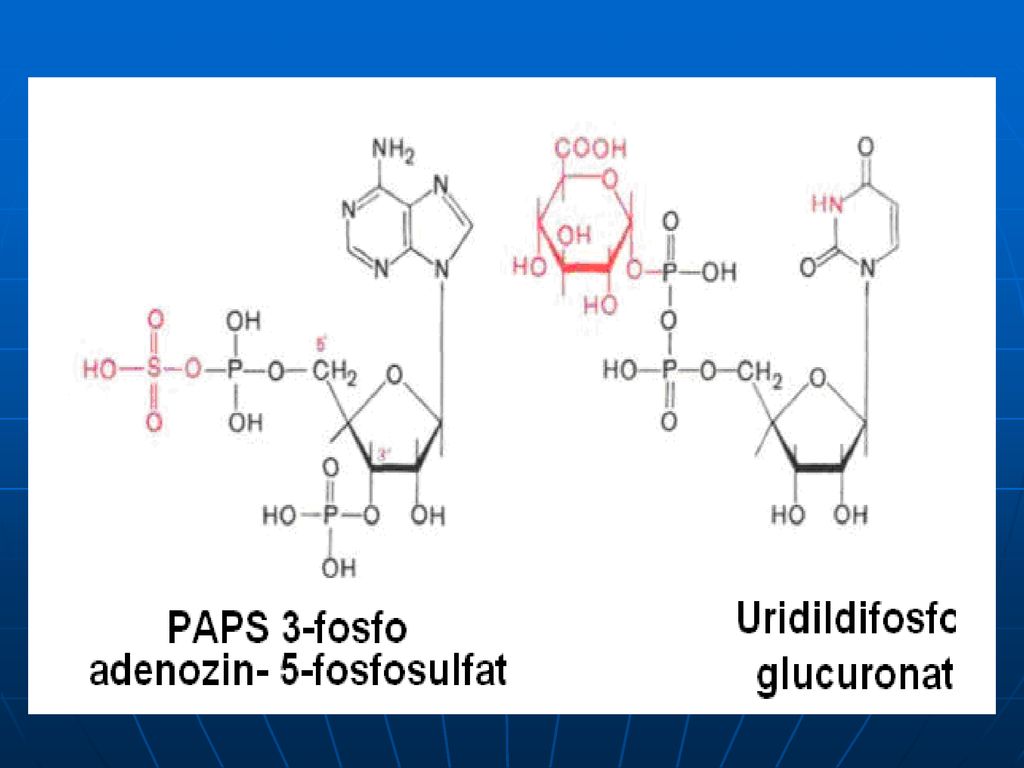
Neutralizarea Aceste produse toxice se absorb din intestin şi sînt neutralizate în ficat. în ficat - în prealabil substanţele toxice sînt oxidate (scatol – scatoxil, indol – indoxil). Ficatul conţine E specifice – arilsulfotransferaza şi UDP – glucoroniltransferaza – ce transferă resturile de acid (a sulfuric sau glucuronic) la substanţele toxice, rezultând compuşi conjugaţi netoxici, eliminaţi prin urină. Menţionăm că resturile de acid sunt în formele active: A sulfuric – PAPS - 3’ –fosfoadenozin –5’fosfosulfat A glucuronic - UDP-glucuronat scatoxil + UDP-glucuronat---scatoxilglucuronat + UDP indoxil + PAPS indoxilsulfat + PAP
. Ficatul conţine E specifice – arilsulfotransferaza şi UDP – glucoroniltransferaza – ce transferă resturile de acid (a sulfuric sau glucuronic) la substanţele toxice, rezultând compuşi conjugaţi netoxici, eliminaţi prin urină. Menţionăm că resturile de acid sunt în formele active: A sulfuric – PAPS - 3’ –fosfoadenozin –5’fosfosulfat. A glucuronic - UDP-glucuronat. scatoxil + UDP-glucuronat---scatoxilglucuronat + UDP. indoxil + PAPS --- indoxilsulfat + PAP.")
42
Sarea de potasiu a indoxilsulfatului se numeşte indican.
Cantitatea de indican din urină indică gradul de putrefacţie în intestin şi starea funcţională a ficatului.

43
Proba Kwick Metodă de apreciere a funcţiei de detoxifiere a ficatului
Se administrează 4 g benzoat de Na, care conjungîndu-se în ficat cu glicină formează acid hipuric eliminat cu urina. Dacă funcţia de barieră a ficatului este normală, peste 6 ore în urină se determină nu mai puţin de 3,6 g acid hipuric

44
Soarta aminoacizilor absorbiţi. AA:
Participă la formarea fondului metabolic comun al AA care vor fi utilizaţi pentru: sinteza proteinelor sinteza glucidelor sinteza lipidelor sinteza hormonilor sinteza de baze azotate purinice, pirimidinice sinteza hemului Sinteza neurotranslatorilor Sinteza porfirinelor Sinteza anserinei, carnozinei Formarea aminelor biogene

45
Transportul aminoacizilor în celule.
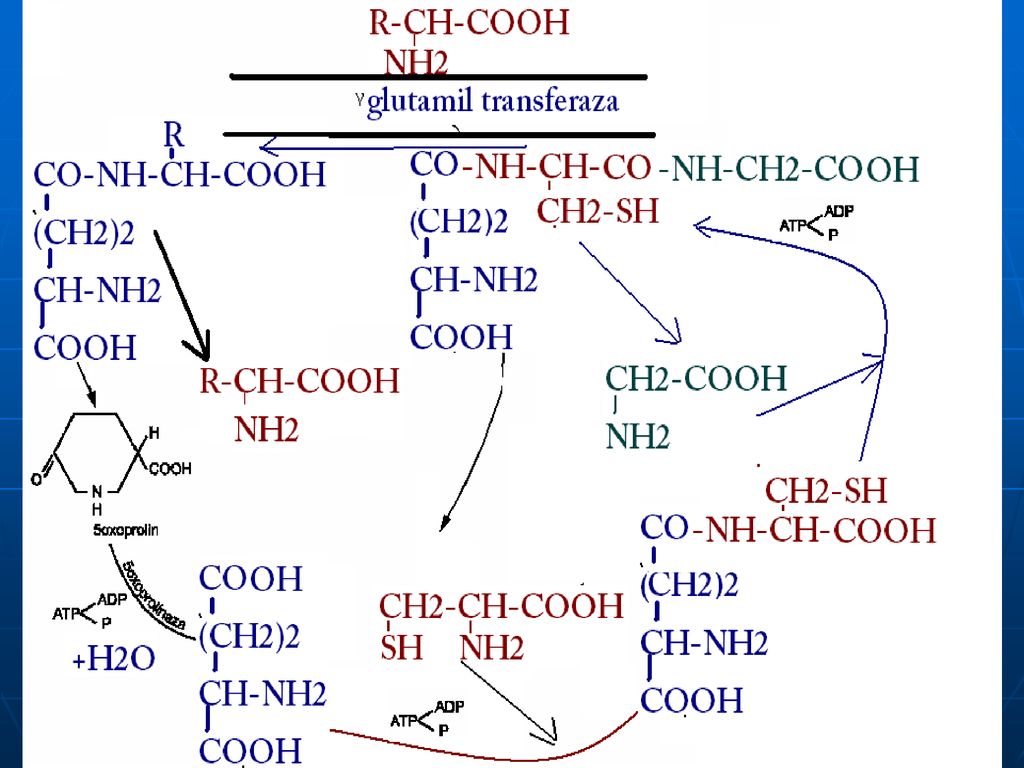
Se realizează cu ajutorul: Transportorilor membranari (reglaţi de insulină) ciclului Glutamil Transferazic (activ în intestin, creier, rinichi, glande salivare) E- glutamiltransferaza Co - glutationul
 ciclului Glutamil Transferazic (activ în intestin, creier, rinichi, glande salivare) E- glutamiltransferaza. Co - glutationul.")
47
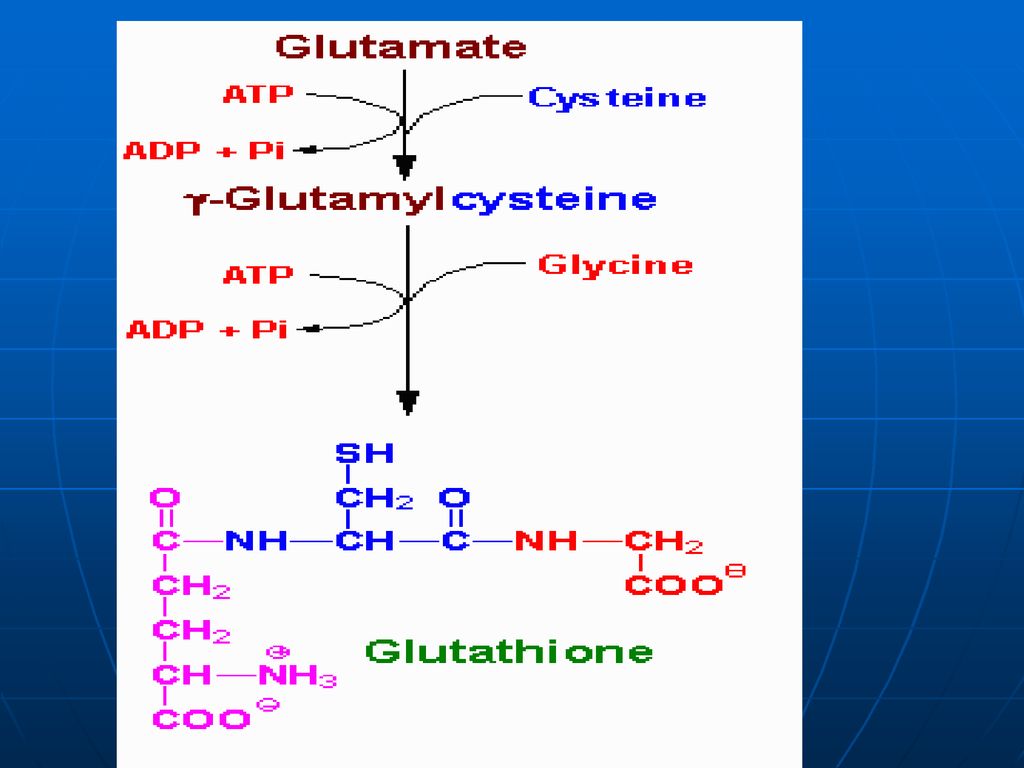
Glutamatul Aminoacid în afara celulei Membrana citoplasmatică
Gama- Glutamil Transferaza Translocaza Gama Glutamil Cisteinil Glicina Cisteinil Glicina Purtător Gama Glutamil AA Dipeptidaza Glutation sintetaza -GlutamilcicloTrasferaza Glicina Gama Glutamil Cisteina Eliberarea AA AA în celulă Cisteina -Glutmil Cisteinil Sintetaza 5-Oxoprolina 5 oxoprolin Glutamatul

48
Căile generale de catabolizare a AA

49
Obiectivele: Soarta aminoacizilor absorbiţi. Transportul aminoacizilor în celule. Metabolizarea NH2-grupelor: Dezaminarea aminoacizilor. Tipurile. Glutamatdehidrogenaza. Transaminarea aminoacizilor. Aminotransferazele şi importanţa clinică a determinării activităţii transanminazelor. Dezaminarea indirectă a aminoacizilor. Decarboxilarea aminoacizilor. Influenţa aminelor biogene asupra funcţiilor fiziologice ale organismului. Detoxifierea aminelor biogene. Metabolizarea -cetoacizilor rezultaţi din aminoacizi. Detoxifierea amoniacului: sinteza glutaminei, carbamoilfosfatului, aminarea reductivă a -cetoglutaratului. Biosinteza ureei. Importanţa clinică a determinării ureei în sînge şi în urină. Biosinteza aminoacizilor neesenţiali în organismul animal.

50
Căi generale şi particulare de catabolizare a AA
căile de degradare, legate de transformarea grupărilor NH2; decarboxilarea gr. α COOH ale a/a – cu formarea aminelor biogene. căile de degradare a scheletelor de atomi de carbon ale a/a;

51
Căile generale de catabolizare pot fi divizate în următoarele grupe:
Dezaminarea. Transaminarea Decarboxilarea

52
Dezaminarea – scindarea grupelor NH2 din poziţia ale AA sub formă de NH3

53
reductivă hidrolitică intramoleculară oxidativă
Sunt posibile 4 tipuri de dezaminare: reductivă +2H R-CH-COOH -----→ R-CH2-COOH + NH3 ׀ NH2 hidrolitică +H2O R-CH-COOH → R-CH-COOH + NH3 ׀ ׀ NH OH intramoleculară R-CH2-CH-COOH → R-CH=CH-COOH + NH3 oxidativă +1/2O2 R-CH-COOH → R-C-COOH + NH3 ׀ ׀׀ NH O

54
Dezaminarea oxidativă
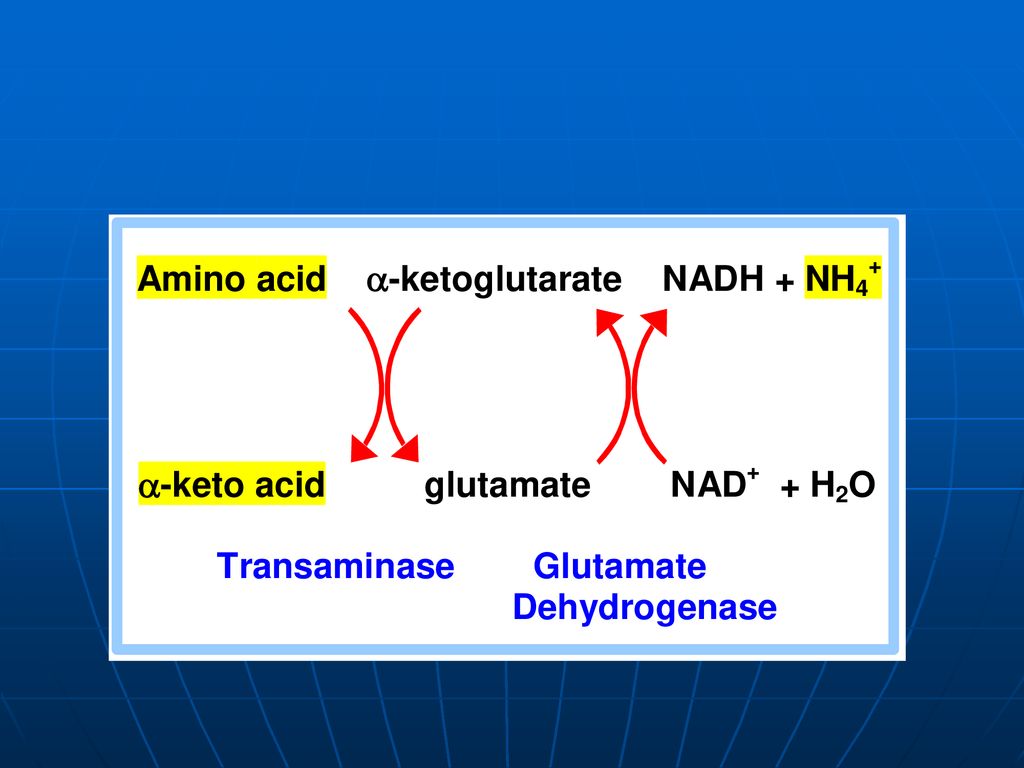
Pentru majoritatea organismelor, inclusiv omul şi animalele, este caracteristică – DO directă indirectă - transdezaminare a. transaminare b. Dezaminarea acidului glutamic

55
DO directă E – oxidazele Co -L-AA → FMN şi FAD;D-AA → FAD
FADH2+O FAD+H2O2 H2O H2O+1/2O2

56
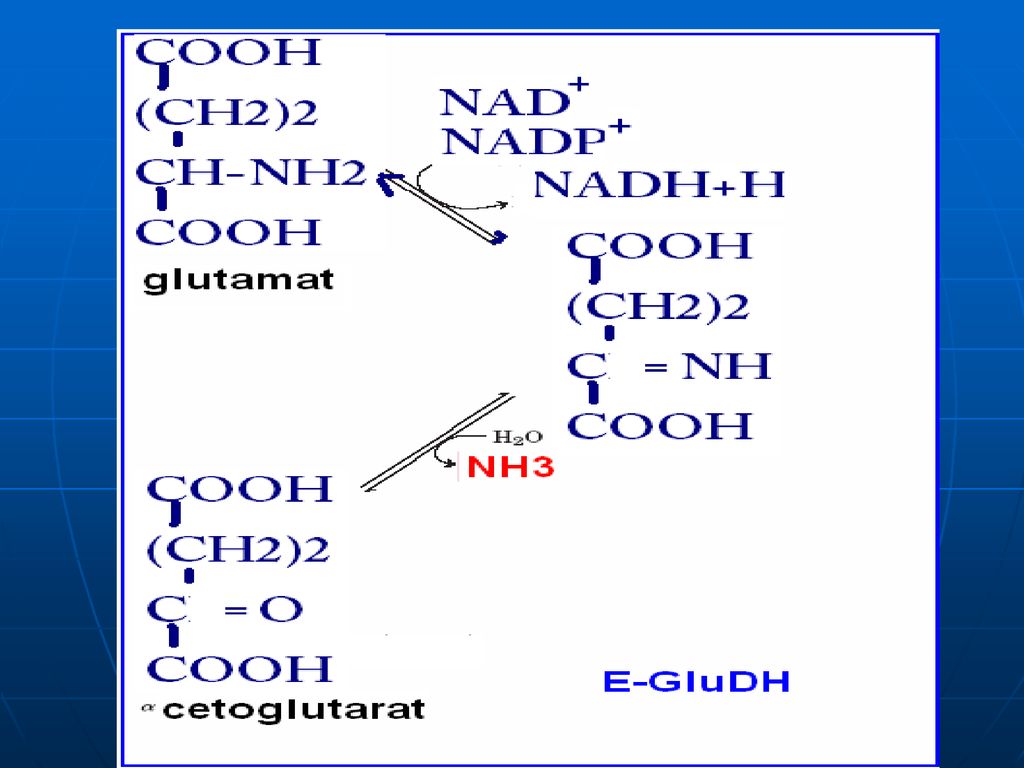
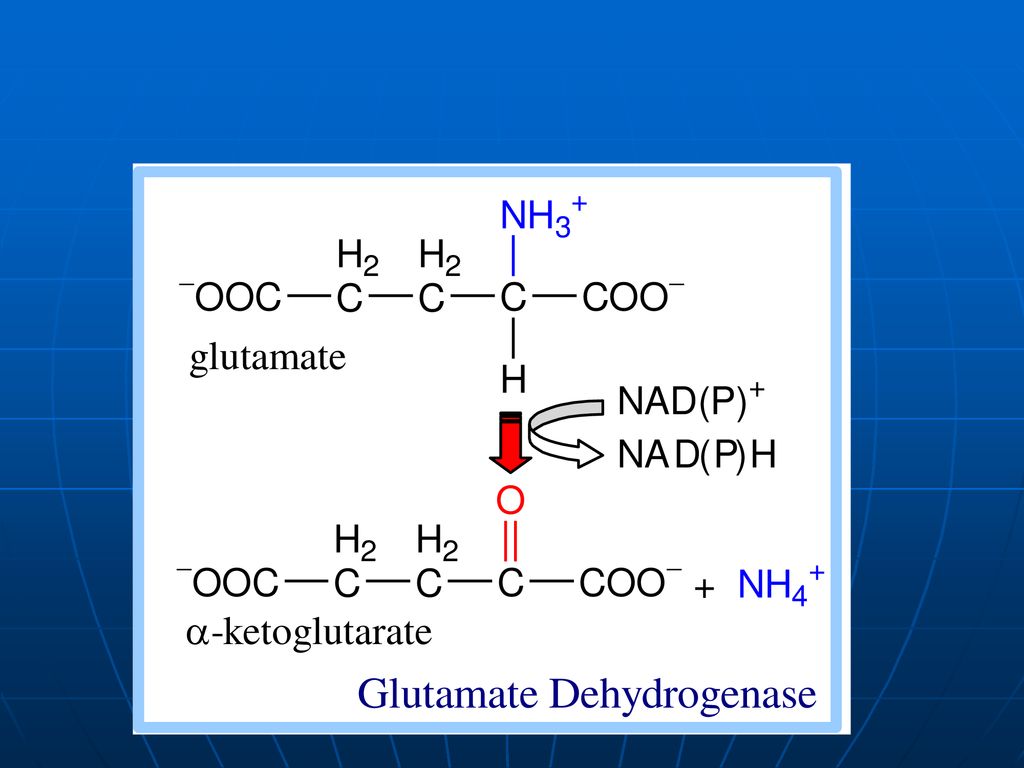
DO a AA În ţesuturi la pH fiziologic e activă numai oxidaza D-AA, pe când toţi AA alimentari (ţesuturile org.) => L AA. pH optim pentru L oxidaze => pH=10,0 => în condiţii fiziologice e activă numai L-enzima, ce catalizează dezaminarea oxidativă a a. glutamic => glutamatdehidrogenaza (enzima anaerobă). Co - NADP+ , NAD+ GluDH =>compusă din 6 subunităţi Activatori: ADP, GDP inhibitori: ATP, GTP
. Co - NADP+ , NAD+ GluDH =>compusă din 6 subunităţi. Activatori: ADP, GDP inhibitori: ATP, GTP.")
58
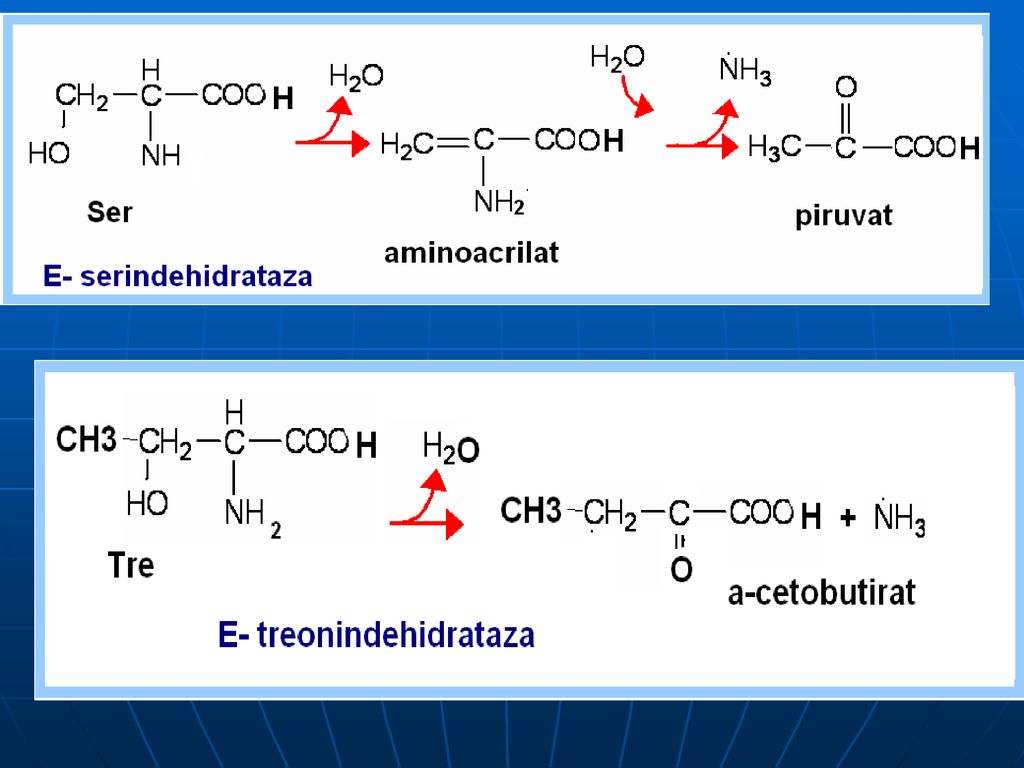
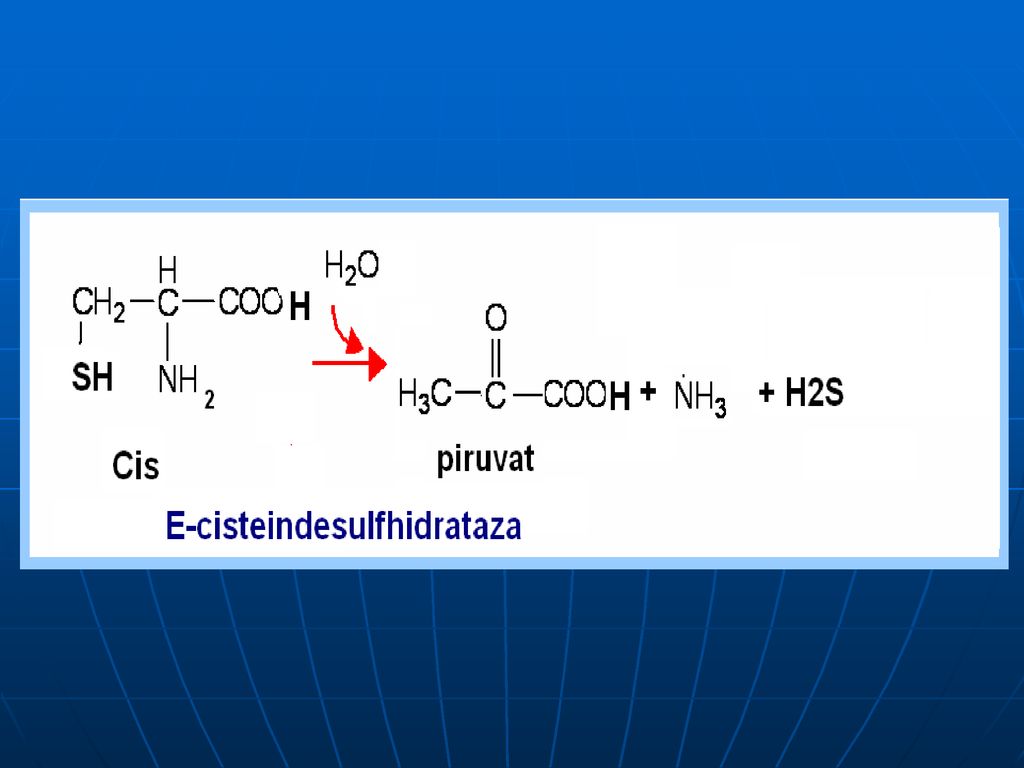
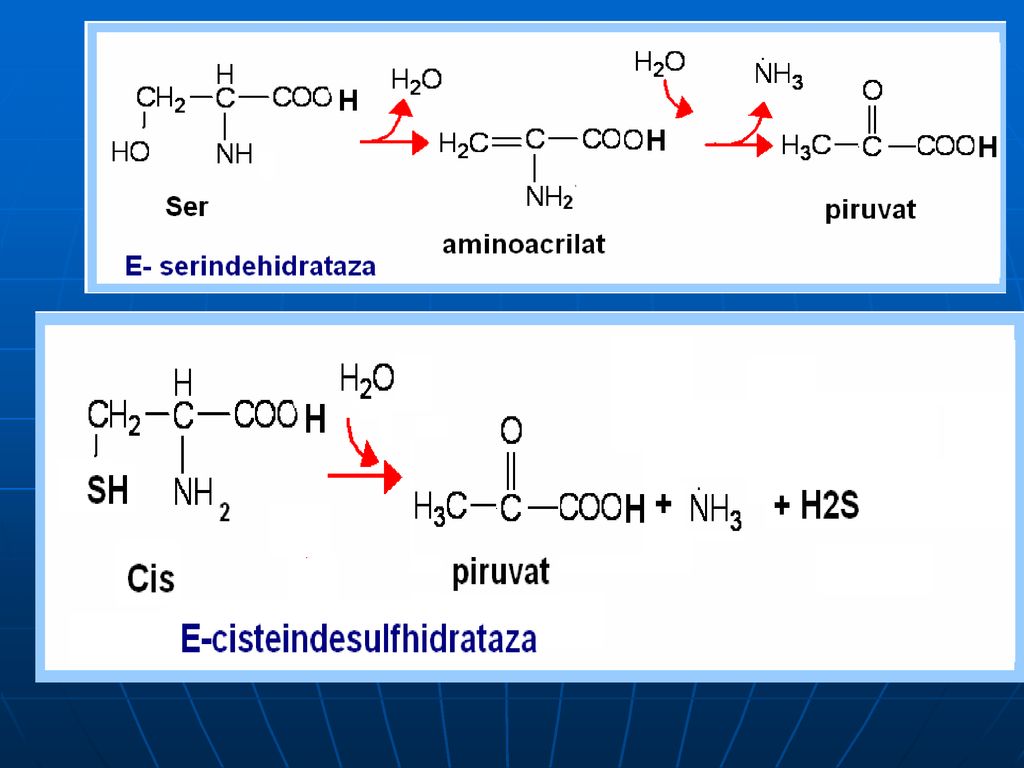
Oxidativ direct în organism sunt dezaminaţi 3 AA: Ser, Tre (E- dehidrataze; Co- B6), Cis (E- desulfhidratază)
, Cis (E- desulfhidratază)")
61
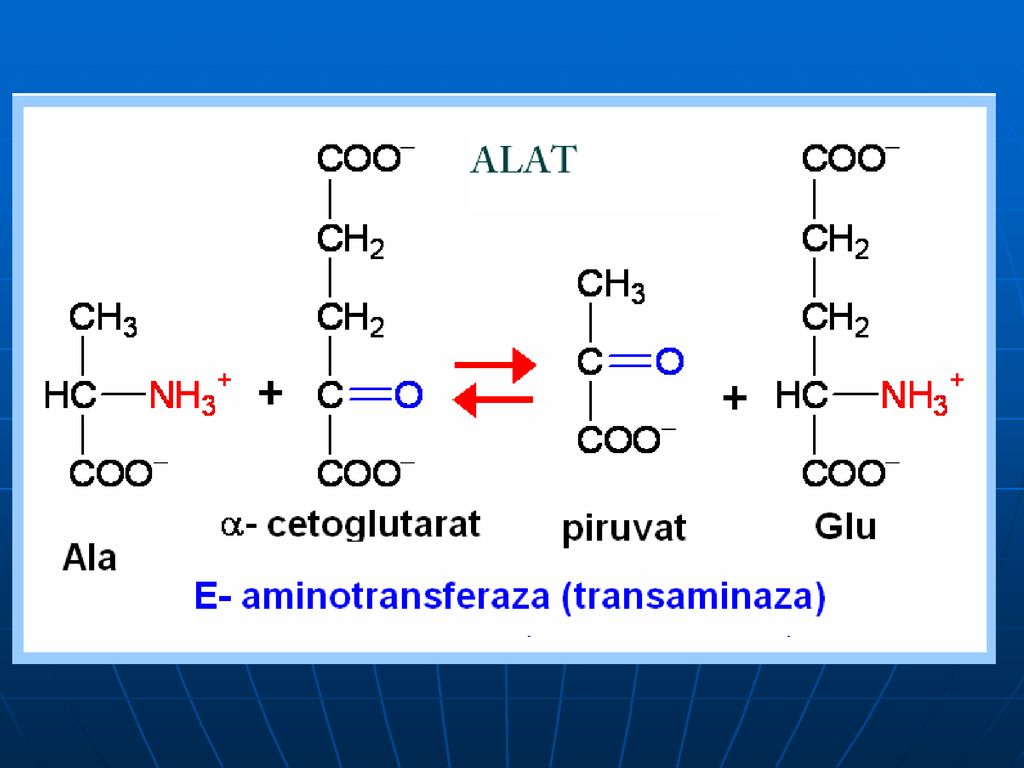
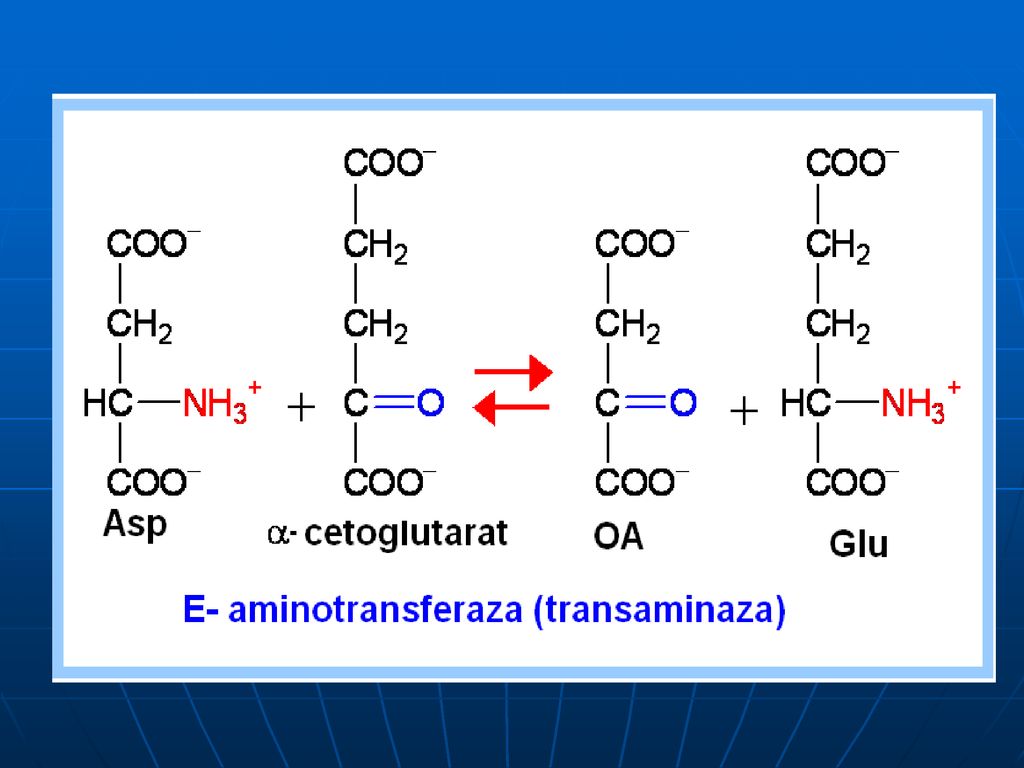
Transaminarea este transferul aminogrupei de la orice AA la α-cetoacid, cu formarea unui nou AA şi nou cetoacid fără formarea de NH3. sunt reacţii reversibile; E → transaminaze (aminotransferaze); coenzime piridoxalfosfat (PALP) şi piridoxaminfosfatul (PAMP); E manifestă specificitate de grup (utilizând în calitate de S cîţiva AA) excepţie => Liz şi treonina.
; coenzime piridoxalfosfat (PALP) şi piridoxaminfosfatul (PAMP); E manifestă specificitate de grup (utilizând în calitate de S cîţiva AA) excepţie => Liz şi treonina.")
62
În calitate de acceptor de gr.NH2 servesc 3 cetoacizi:
α-cetoglutaratul => Glu piruvat → Ala; OA → Asp

65
Alaninaminotransferaza (ALAT sau GPT- glutamic piruvic transaminază)
Aspartataminotransferaza (ASAT sau GOT – glutamic-OA transaminază) Creşterea nivelului seric al lor este cauza leziunilor celulare la nivelul ţesutului afectat (sd de citoliză a ţesuturilor în care se află aceste E) ALAT – se află în faza solubilă a celulei şi în C % mult mai mari în hepatocite (raportul nivelul hepatic/nivelul extrahepatic: 10/1) ASAT – ficat, inimă,muşchii sceletici (raportul nivelul hepatic/nivelul extrahepatic: 1/1)
 Creşterea nivelului seric al lor este cauza leziunilor celulare la nivelul ţesutului afectat (sd de citoliză a ţesuturilor în care se află aceste E) ALAT – se află în faza solubilă a celulei şi în C % mult mai mari în hepatocite (raportul nivelul hepatic/nivelul extrahepatic: 10/1) ASAT – ficat, inimă,muşchii sceletici (raportul nivelul hepatic/nivelul extrahepatic: 1/1)")
66
ALAT: ASAT: hepatita infecţioasă;
hepatite antiicterice - perioada de incubare; hepatopatie toxică; hepatita cronică. în ciroza ficatului şi icterul mecanic cresc puţin. ASAT: ↑ infarct miocardic în 95%; ↑raportului DE RITTIS (GOT/GPT, normă 1,33) ↑ activ. sale apare peste 4-6 ore, manifestându-se celor ore; după 3-7 zile => activitate atinge valori normale.
 ↑ activ. sale apare peste 4-6 ore, manifestându-se celor ore; după 3-7 zile => activitate atinge valori normale.")
67
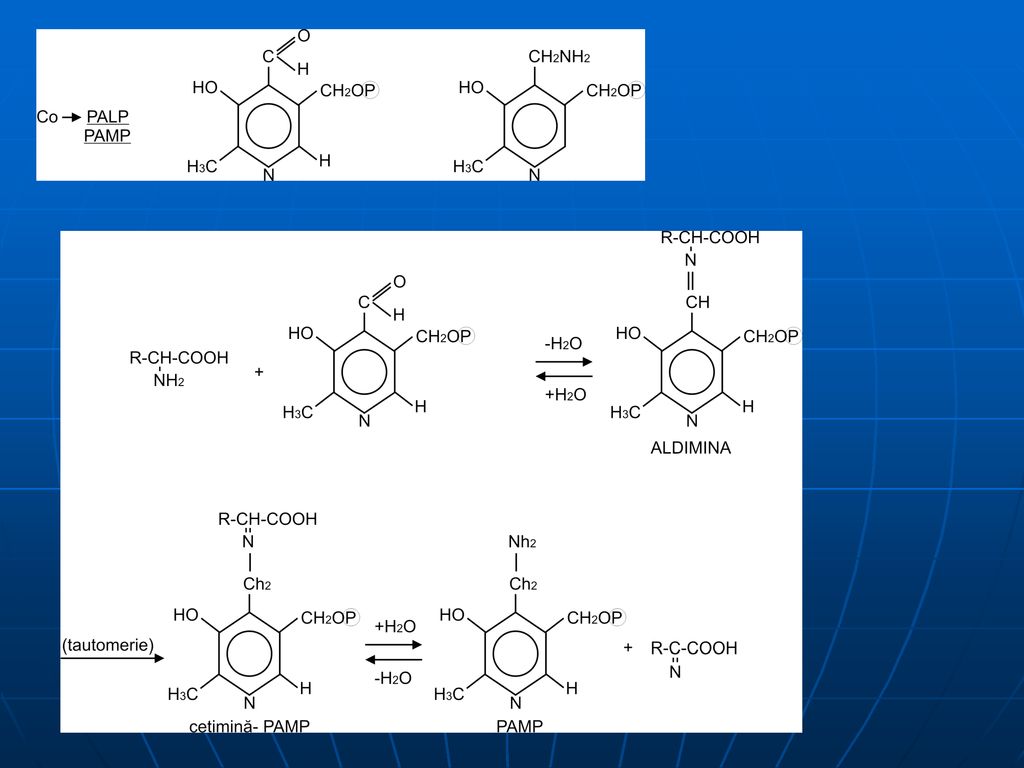
Mecanismul transaminării
1. Legarea PALP de un rest de Lys al E (compus de tip bază Schiff)
")
68
2. PALP reacţionează cu AA, formând o bază Schiff, care se detaşează de molecula E
3. deplasarea dublei legături (aldemina –cetimină), eliberarea alfa-cetoacidului şi formarea intermediară a PAMP
, eliberarea alfa-cetoacidului şi formarea intermediară a PAMP.")
69
Mecanismul transaminării
Prima etapă

70
Mecanismul transaminării
A doua etapă

72
Sensul biologic al reacţiilor de transaminare
constă în „adunarea”sau “colectarea” gr.NH2 ale tuturor AA în structura moleculei de acid glutamic. Glu => pătrunde în mitocondrii => dezaminarea propriu zisă a acidului glutamic E – glutamatdehidrogenaza (GluDH) Co - NADP+ , NAD+
 Co - NADP+ , NAD+")
74
Reacţia de transdezaminare
În prima etapă toţi AA întră într-o reacţie de transaminare cu -cetoglutaratul, rezultînd Glu - localizat în citoplasmă. Glu este dezaminat cu participarea enzimei glutamatdehidrogenaza (GluDH) - mitocondrii
 - mitocondrii.")
76
Soarta α cetoacizilor rezultaţi din AA
Biosinteza AA dispensabili-transreaminare (sinteza AA din α cetoacizii corespunzători) Biosinteza Gl şi glicogenului Biosinteza AG şi lipidelor Ciclul Krebs – pînă la CO2 şi H2O
 Biosinteza Gl şi glicogenului. Biosinteza AG şi lipidelor. Ciclul Krebs – pînă la CO2 şi H2O.")
77
Sinteza AA neesenţiali
din intermediarii de degradare a glucidelor: glutamat + piruvat -cetoglutarat + alanina 3-fosfoglicerat serina glicină ribozo-5-fosfat –fosforibozilpirofosfat--His din metaboliţii ciclului Krebs prin transreaminare 1. -cetoglutarat + NH glutamat 2. glutamat + oxaloacetat----- -cetoglutarat + aspartat din aminoacizi esenţiali O2+ NADPH2 Phe Tyr+ NADP+ H2O Met Cys

78
Ficat Proteinele celulare AA Ceto acizii Ala din muşchi Glu din muşchi
şi alte ţesuturi

79
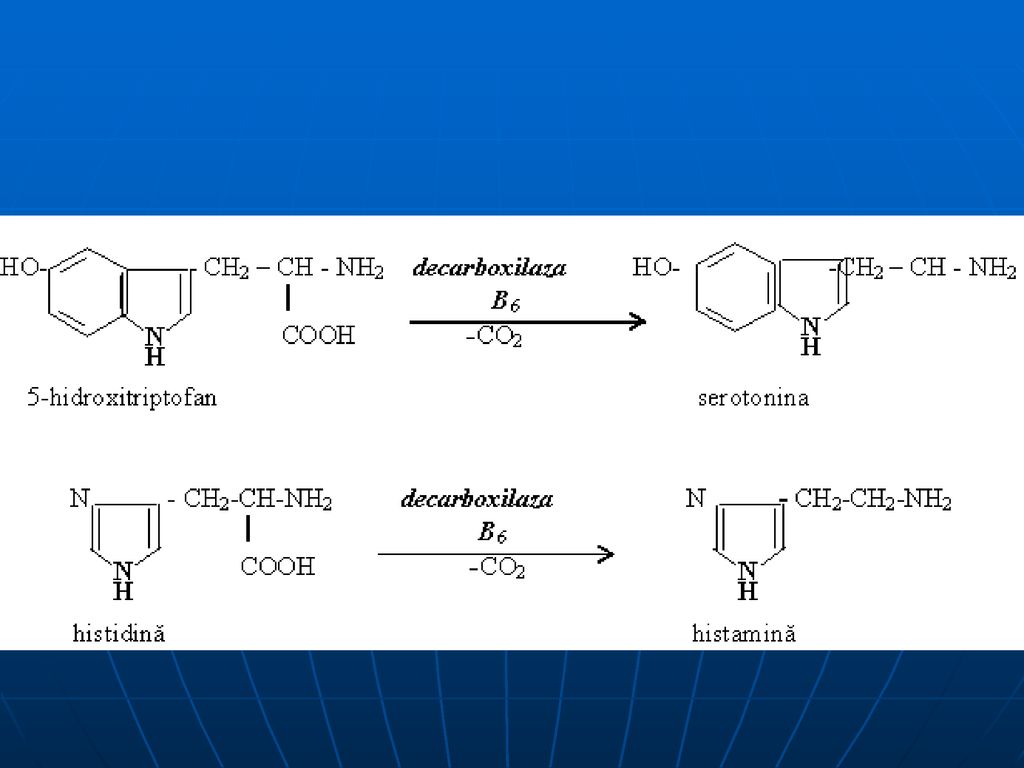
Decarboxilarea AA scindarea CO2 de la gr. α-carboxil a AA cu formarea de amine biogene. E- decarboxilaza (Co- PALP)
 .")
82
aminele biogene Triptofan → triptamină 5 oxotriptofan → serotonină
3, 4 dioxifenilalanină → dofamină histidina → histamină glutamatul → γ aminobutirat

85
GLU ▬►gamma-aminobutitar (GABA)
")
86
Rolul aminelor biogene
Serotonina – mediator chimic, vasoconstrictor: la reglarea TA t corpului Respiraţiei filtraţiei renale este mediator al SNC participă în dezvoltarea alergiei, toxicozei în timpul gravidităţii, diatezelor hemoragice. Dofamina → sinteza catecolaminelor Histamina: vazodilatator, ↑ secreţia HCl, participă în reacţiile de sensibilizare şi desensibilizare a organismului. γ aminobutiratul – efect inhibitor în substanţa cenuşie a creierului. Se utilizează pentru tratarea afecţiunilor sistemului nervos, provocate de excitaţii excesive.

87
Neutralizarea aminelor biogene
E– mono- sau diaminooxidazele Proces ireversibil 2 etape: R-CH2-NH2 + E-FAD+ H2O →R-COH+ NH3 + E-FADH2 E-FADH2 + O2 → E-FAD + H2O2 2H2O2 → H2O +O2

88
Soarta amoniacului NH3 se formează în următoarele procese:
dezaminarea AA; detoxifierea aminelor biogene; degradarea BA purinice şi pirimidinice; dezaminarea amidelor AA (Asn, Gln); Putrefacţia AA în intestinul gros sub acţiunea microflorei
; Putrefacţia AA în intestinul gros sub acţiunea microflorei.")
89
NH3 – o combinaţie toxică, îndeosebi pentru celulele nervoase.
Efectul toxic se exprimă prin C% mare de ioni de amoniu, ce dezechilibrează reacţia catalizată de GluDH, cu formarea Glu (o transformare excesivă). Această cauzează epuizarea α-cetoglutaratului (produs intermediar al ciclului Krebs) – cu reducerea reacţiilor de generare a ATP. Carenţa energetică conduce la micşorarea sintezei mediatorilor nervoşi şi dereglarea transmiterii impulsului – blocarea funcţiei SNC. α-cetoglutarat + NH4 + NADPH + H+ → glutamat +NADP
. Această cauzează epuizarea α-cetoglutaratului (produs intermediar al ciclului Krebs) – cu reducerea reacţiilor de generare a ATP. Carenţa energetică conduce la micşorarea sintezei mediatorilor nervoşi şi dereglarea transmiterii impulsului – blocarea funcţiei SNC. α-cetoglutarat + NH4 + NADPH + H+ → glutamat +NADP.")
90
Căile de neutralizare a NH3
1.În ţesuturi(muşchi, creier, glande): are loc sinteza glutaminei sub acţiunea glutaminsintetazei citoplasmatice (ATP şi Mg) → Glu + NH3 + ATP → Gln + ADP + Pa - proces ireversibil Gln – sânge – ficat şi rinichi c% Gln în sînge - de 3-5 ori mai mare faţă de alţi AA
: are loc sinteza glutaminei sub acţiunea glutaminsintetazei citoplasmatice (ATP şi Mg) → Glu + NH3 + ATP → Gln + ADP + Pa. - proces ireversibil. Gln – sânge – ficat şi rinichi. c% Gln în sînge - de 3-5 ori mai mare faţă de alţi AA.")
91
2. În ficat şi rinichi:Gln (sub acţiunea glutaminazei mitocondriale):
Gln +H2O => Glu + NH3 –proces ireversibil Aceste 2 etape împreună - ciclul glutamină-glutamic În ficat % din conţinutul total de NH3 - sinteza ureei. În tubii renali NH3 este neutralizat cu formarea sărurilor de amoniu. NH3 + H+ + Cl- → NH4Cl

92
Ciclul Ala-Gl În muşchi: AA (prin dezaminare oxidativă)- NH3
NH3+alfa-cetoglutarat▬►Glu (GDH) Glu+Piruvat ▬► alfa-cetoglutarat +Ala În sânge: Ala ▬► în ficat În ficat: Ala + alfa-cetoglutarat ▬►Piruvat+Glu (GDH—NH3—uree) Piruvatul prin gluconeogeneză --- Gl Gl în sânge ▬►muşchi ▬►piruvat
 Glu+Piruvat ▬► alfa-cetoglutarat +Ala. În sânge: Ala ▬► în ficat. În ficat: Ala + alfa-cetoglutarat ▬►Piruvat+Glu (GDH—NH3—uree) Piruvatul prin gluconeogeneză --- Gl. Gl în sânge ▬►muşchi ▬►piruvat.")
93
Sinteza ureei (Krebs-Henseleit) ciclul ornitinic sau ureogenetic
în mitocondrii: Sinteza carbomoil fosfatului E –carbomoilfosfatsin-tetază (E biotinică, modulată pozitiv de N-acetilglutamat)
")
94
2. Transferul carbomoilfosfatului pe ornitină- citrulinei
E- ornitin-carbomoil-transferază

95
În citozol: Condensarea citrulinei cu Asp

97
E- Arginaza: Activată- Co, Mn Inhibată- ornitină şi Lyz

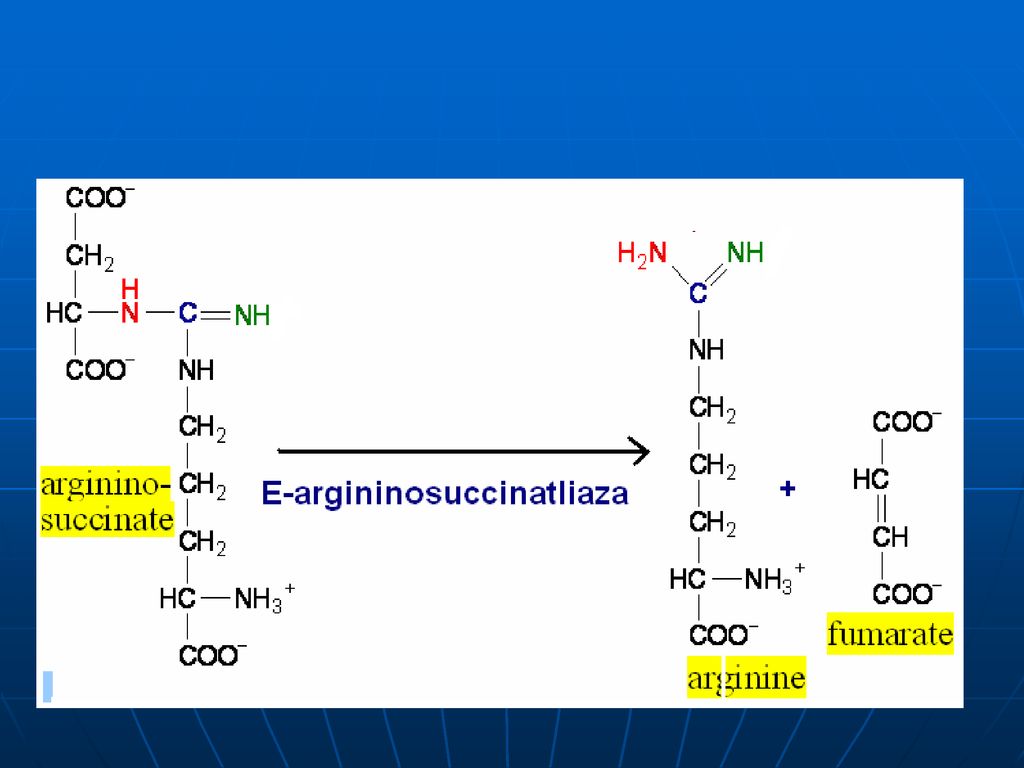
98
Arginaza Ureea ATP Citrulin Ornitin ArgininoSuccinat Aspartat
Ornitin Carbamoil Transferaza Citrulin Ornitin ArgininoSuccinat Sintetaza Aspartat Arginaza Ureea ATP Arginino Succinat Liaza Arginina ArgininoSuccinat

99
Stoichiometria procesului
CO2+NH3+3ATP+Asp+2H2O Urea+2ADP+2Pi+AMP+PPi+fumarat Pentru sinteza ureei sunt necesare 4 legături macroergice fosfat Ureea este netoxică – se elimină prin urină (15-30g/24 ore)- variază proporţional cu cantitatea de proteine îngerate
- variază proporţional cu cantitatea de proteine îngerate.")
100
Relaţia ciclul ornitinic- ciclul Krebs
Ciclul ornitinic e dependent energetic şi metabolic de ciclul Krebs: Energetic – sinteza ATP în ciclul Krebs şi consumul lui în ciclul ornitinic Metabolic – fumarat (se include în ciclul Krebs) ----OA OA---prin transaminare --- Asp Asp---ureogeneză
 ----OA. OA---prin transaminare --- Asp. Asp---ureogeneză.")
102
Urea Cycle Enzymes in mitochondria: 1. Ornithine Trans- carbamylase
Enzymes in cytosol: 2. Arginino- Succinate Synthase 3. Arginino- succinase 4. Arginase.

104
METABOLISMUL INTERMEDIAR AL UNOR AMINOACIZI

105
OBIECTIVELE Metabolismul fenilalaninei, tirozinei şi triptofanului. Rolul acestor aminoacizi în sinteza altor compuşi. Metionina. S-Adenozilmetionina. Rolul acestui aminoacid în organism. Sinteza creatinei. Acidul tetrahidrofolic. Rolul lui în sinteza serinei, metioninei, glicinei, timinei. Metabolismul glicinei, serinei şi cisteinei. Metabolismul aminoacizilor dicarboxilici. Glutamina şi rolul ei în organism; glutaminaza rinichilor. Patologia metabolismului proteic. Tulburările congenitale ale metabolismului aminoacizilor.

106
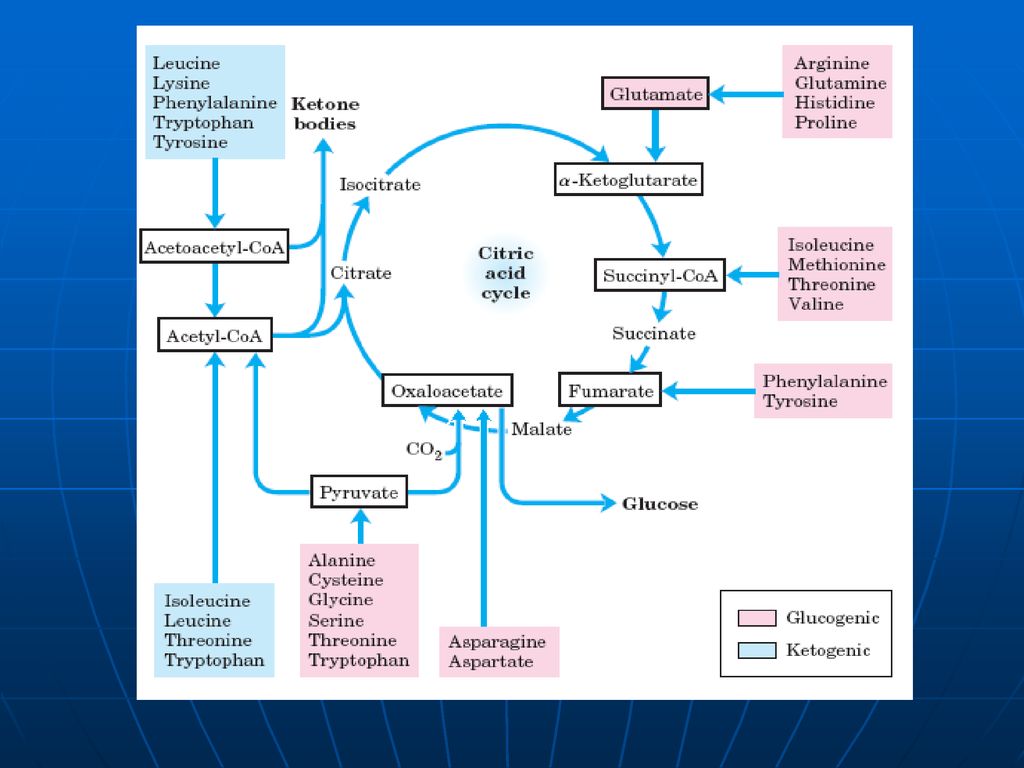
Soarta scheletului de carbon

107
Utilizarea scheletului de carbon al AA
Scheletul de carbon al celor 20AA se modifică în: piruvat, acetil CoA; acetoacetil CoA; OA, alfa-cetoglutarat; succinil CoA; fumarat. AA glucoformatori: servesc pentru sinteza Gl AA cetoformatori: servesc pentru sinteza de lipide şi corpi cetonici. Leu – exclusiv cetogen AA gluco şi cetoformatori: Phe, Tyr, Trp, Ile, Lys

108
Soarta scheletului de carbon

109
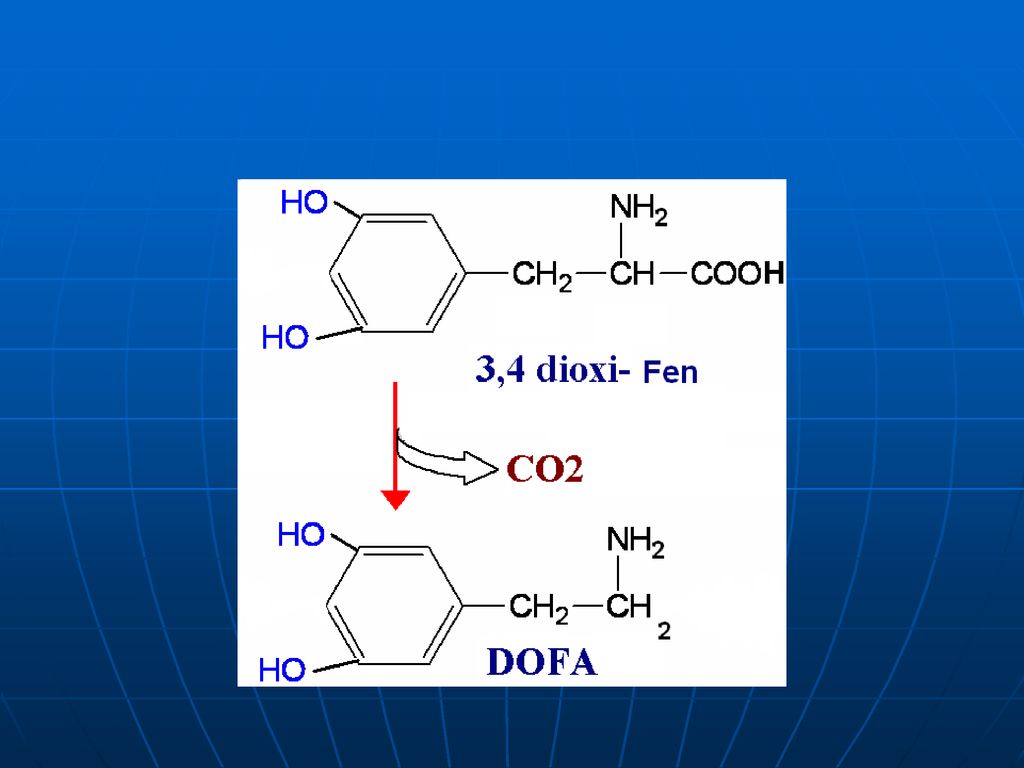
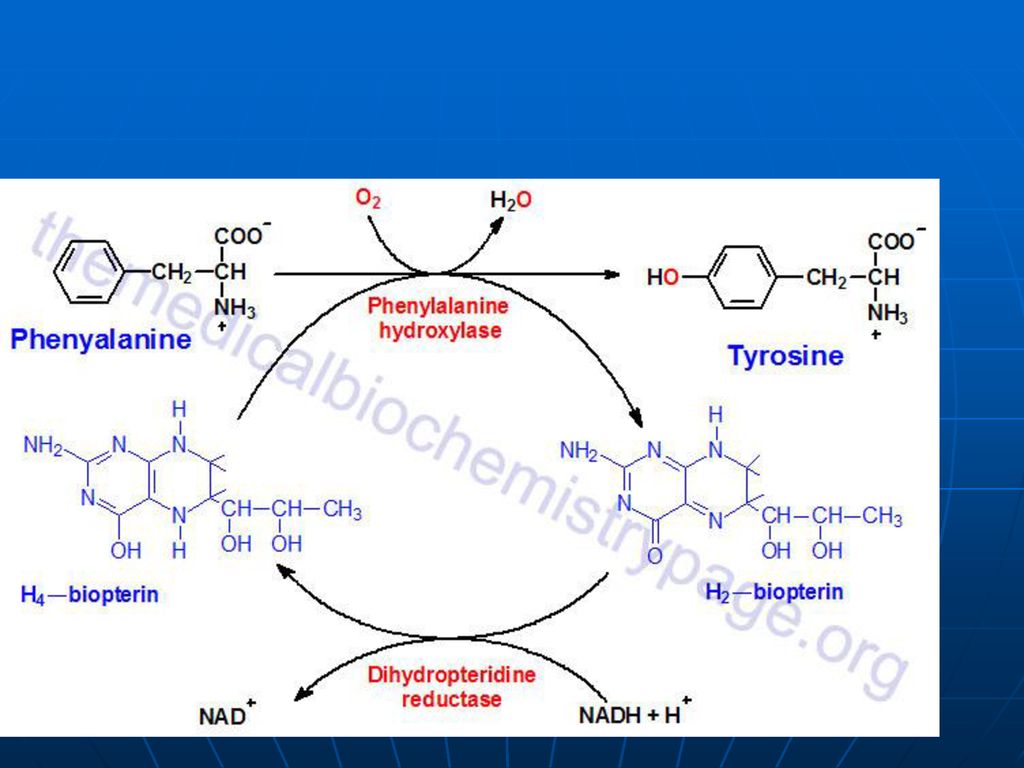
Metabolismul Phe (Fen) şi Tyr A. Sinteza
Fen – AA esenţial Tyr – AA neesenţial- se sintetizează din Fen

111
Lipsa fenilalaninhidroxilazei – fenilcetonurie – acumularea Phe- fenilpiruvat- fenillactat sau fenilacetat (eliminaţi prin urină). În ficat fenilacetat + Gln- fenilacetilGln (urina) Fenilpiruvatul - substanţă toxică în special pentru SNC Retard mental Demielinizări ireversibile Diminuată sinteza DOPA- melaninei; serotoninei. Dietă strictă (până la 6 ani)
")
112
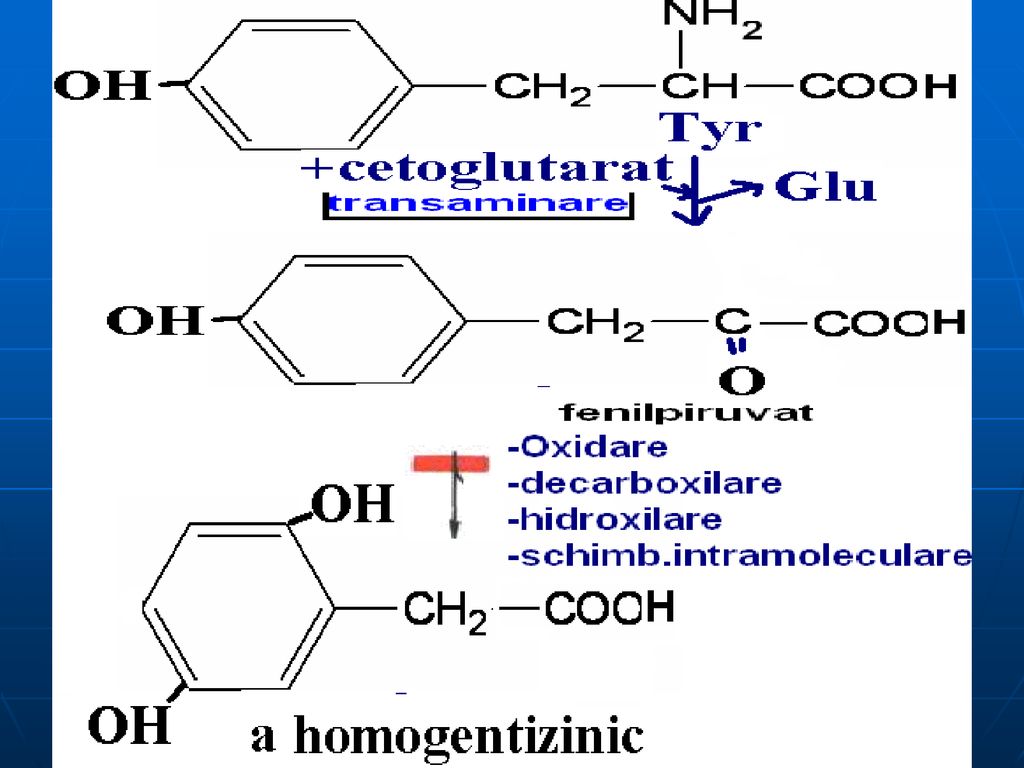
B. Catabolismul Fen şi Tyr
Pînă la fumarat şi acetoacetat

115
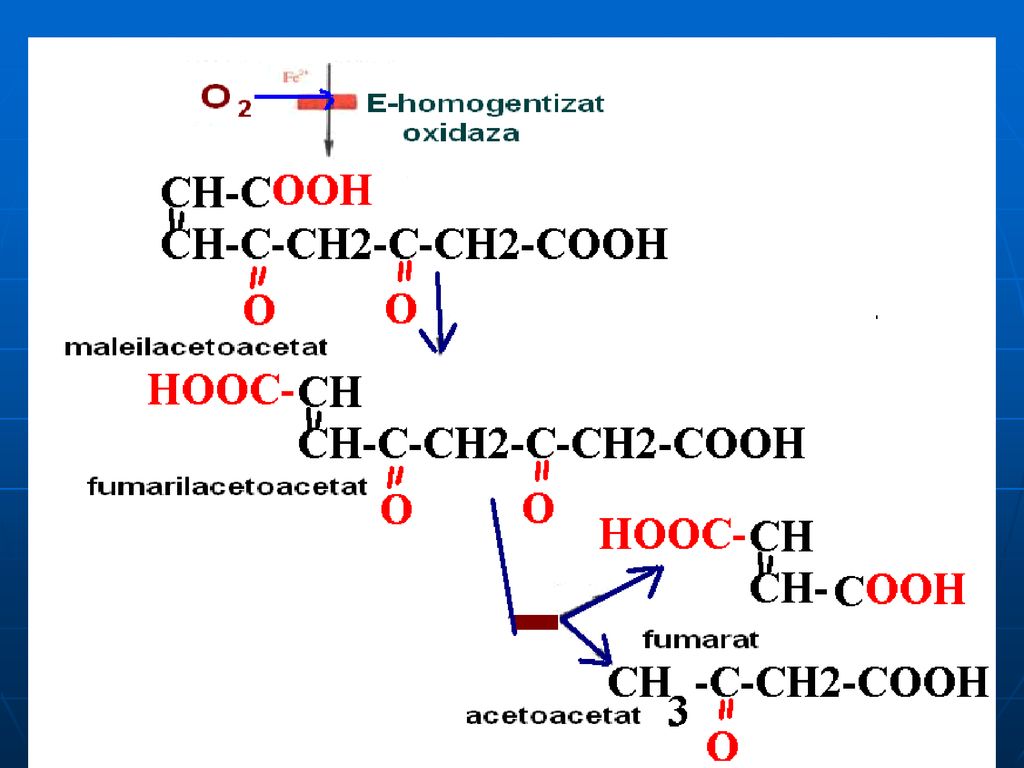
Alcaptonuria - lipsa homogentizinatoxidazei: acumularea a
Alcaptonuria - lipsa homogentizinatoxidazei: acumularea a.homogentizinic în ţesuturi şi eliminarea lui cu urina (urina se colorează în albastru sau negru). Colorarea ţesuturilor (conjunctiv:cartilagiile nasului, urechile se întunecă). Tirozinemia de tip I- lipsa de fumarilacetoacetaza, maleilacetoacetaza: vomă, diaree, deficienţa de creştere (acută- exitus 6-8 luni, cronică – moartea la 10 ani) Tirozinemia de tip II – lipsa Tyr transaminazei ficatului – mărirea c%Tyr, afecţiuni a pielii şi ochiului, retard mintal moderat, dereglarea coordonării mişcărilor Tirozinemia neonatală- deficit de hidroxifenilpiruvathidroxilaza- mărirea c% de Fen şi Tyr în sânge; în urină: Tyr, tiramina, hidroxifenilacetat.
. Colorarea ţesuturilor (conjunctiv:cartilagiile nasului, urechile se întunecă). Tirozinemia de tip I- lipsa de fumarilacetoacetaza, maleilacetoacetaza: vomă, diaree, deficienţa de creştere (acută- exitus 6-8 luni, cronică – moartea la 10 ani) Tirozinemia de tip II – lipsa Tyr transaminazei ficatului – mărirea c%Tyr, afecţiuni a pielii şi ochiului, retard mintal moderat, dereglarea coordonării mişcărilor. Tirozinemia neonatală- deficit de hidroxifenilpiruvathidroxilaza- mărirea c% de Fen şi Tyr în sânge; în urină: Tyr, tiramina, hidroxifenilacetat.")
116
C. Reacţiile metabolice
Din Phe şi Tyr se sintetizează: dopamină Adrenalina, noradrenalina (hormonii medulosuprarenali); Iodtironinele (triiodtironina, tiroxina)- hormonii tiroidei; Melanina (pigmentul organismului); biosinteza proteinelor, enzimelor, unor hormoni, peptidelor biologic active etc.
; Iodtironinele (triiodtironina, tiroxina)- hormonii tiroidei; Melanina (pigmentul organismului); biosinteza proteinelor, enzimelor, unor hormoni, peptidelor biologic active etc.")
117
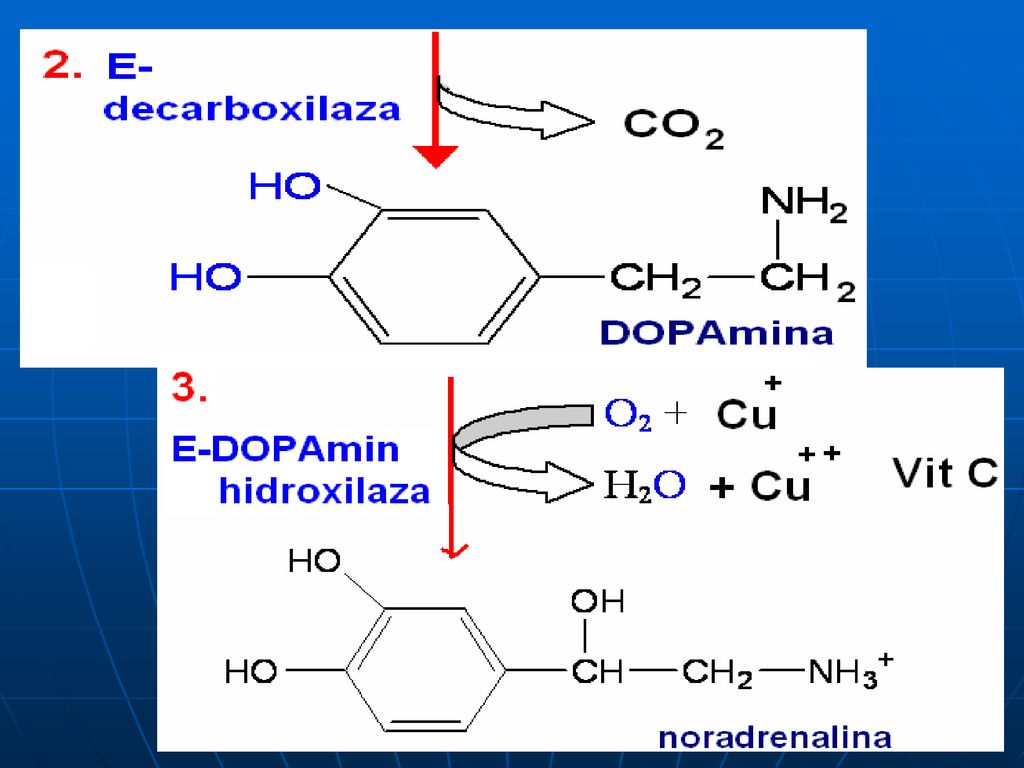
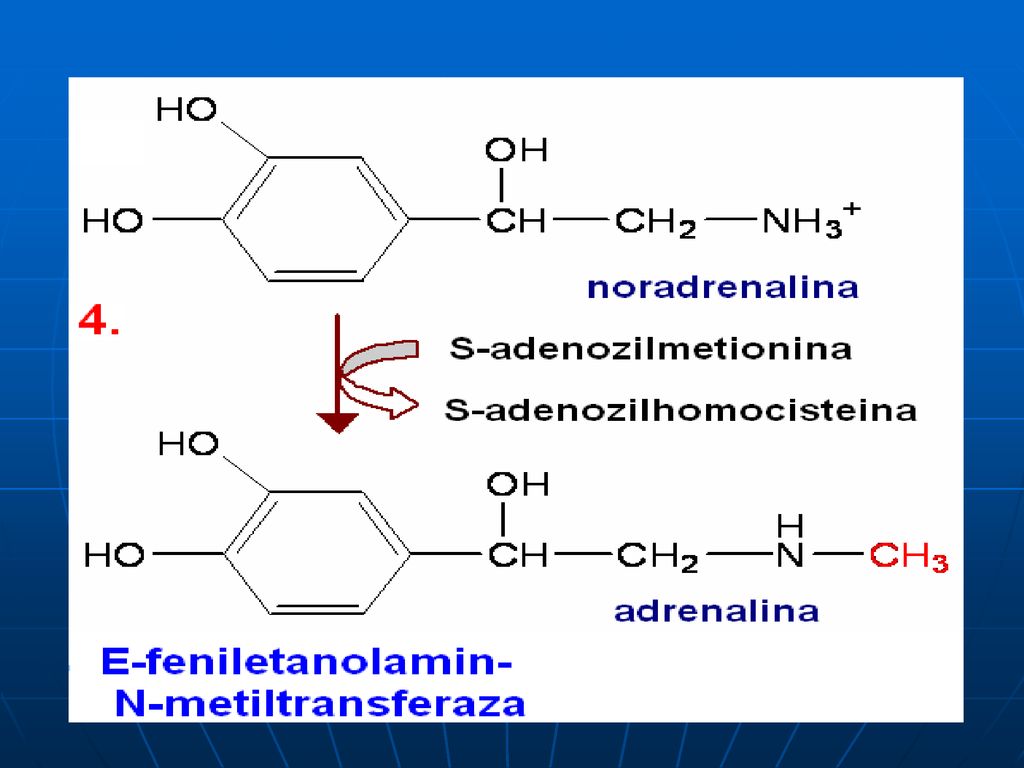
Sinteza catecolaminelor

120
2. Sinteza melaninei

122
Albinismul apare în rezultatul deficienţei echipamentului enzimatic participant la biosinteza melaninei. Bolnavul este lipsit de pigment: alb absolut (pielea şi părul se decolorează) dezvoltaţi mintal normal. Sunt afectaţi de razele solare directe (pielea se afectează, apare hiperemie, ulceraţii etc.)
 dezvoltaţi mintal normal. Sunt afectaţi de razele solare directe (pielea se afectează, apare hiperemie, ulceraţii etc.)")
123
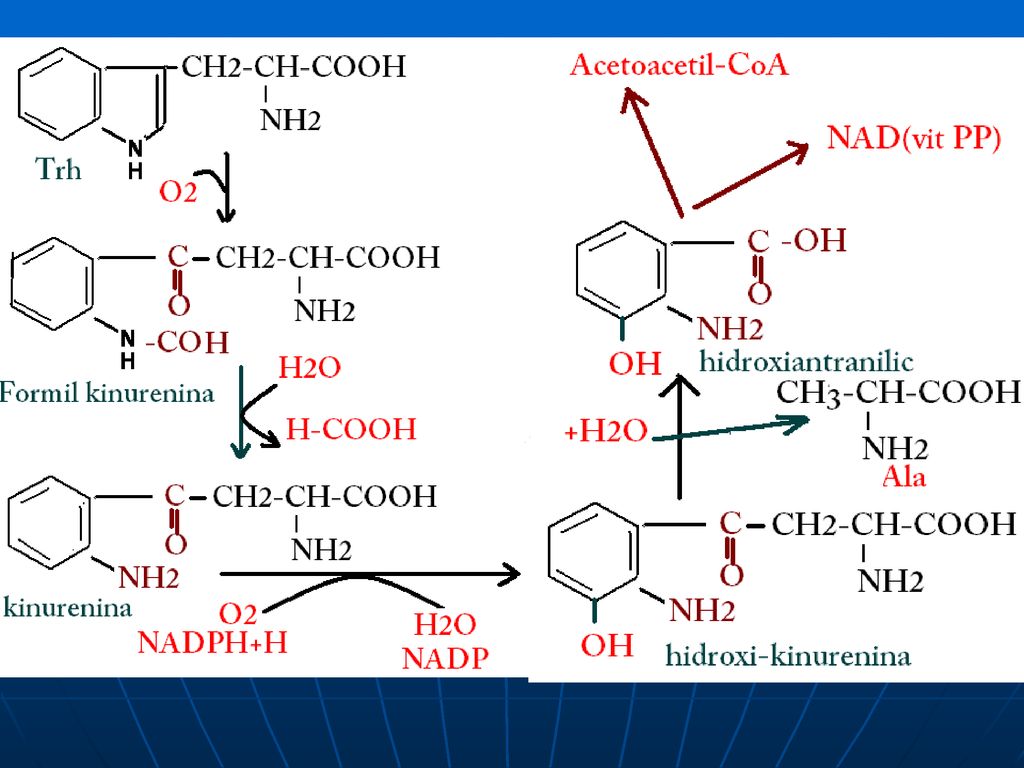
Metabolismul Trh A.Trh – AA esenţial, glucogen şi cetogen
B. Catabolismul Trh: acetoacetil CoA—Acetil CoA în timpul catabolizării produce: Ala - piruvat NAD şi NADP (din hidroxiantranilat)
")
125
Maladia Hartnup Insuficienţa E implicate în catabolismul Trh (tiptofan pirolaza) Uriticărie a pielii Retard mintal, ataxie cerebrală Mătirea c% de Trh şi indolacetic în urină În colon: sub influienţa microflorei bacteriene Trh---- indolilacetic

126
C. Reacţiile metabolice:
sinteza serotoninei sinteza triptaminei sinteza Ala Sinteza NAD

127
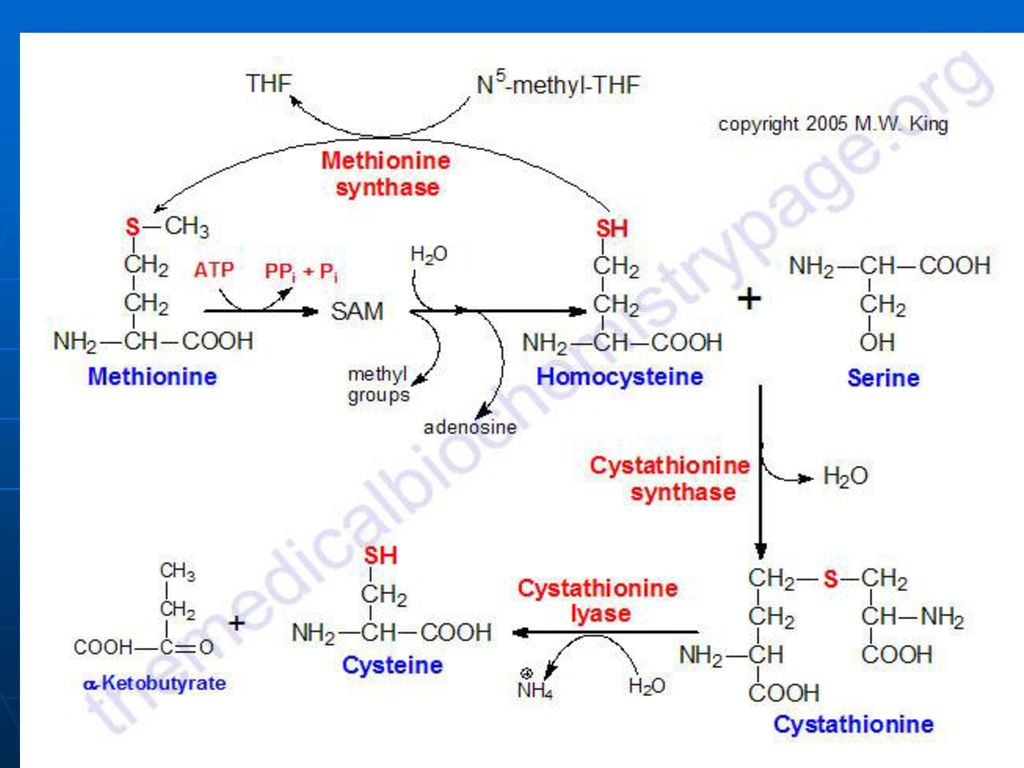
Metionina. S-Adenozilmetionina
A. Met – AA esenţial, glucogen S-adenozil metionina - donor de gruparea CH3

128
B. Catabolismul Met – succinil CoA:
Met---S-adenozilMet---S-adenozil-homocistein--homocistein+adenozin

129
b. Homocisteina+Ser---cistationina
c. Cistationina---NH3, Cis, cetobutirat d. Cetobutiratul---propionil CoA----metilmalonil CoA---succinil CoA

131
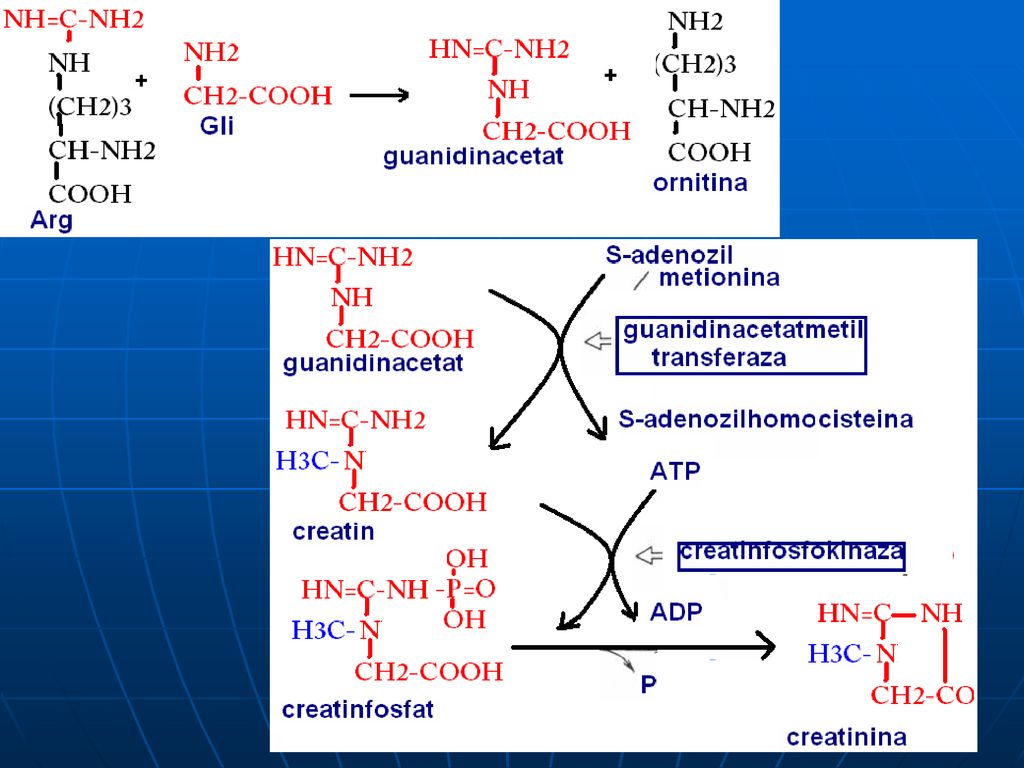
C. Reacţiile metabolice
S-adenozil-Met participă la sinteza: Fosfatidilcolinei Adrenalinei Creatinei La metilarea BA purinice şi pirimidinice: N1-metiladenozin, metilguanozin (N2,N7)
")
133
Creatinfosfatul – singurul compus cu legături macroergice pe care organismul îl poate depozita în muşchi. La un efort fizic se eliberează ATP mai rapid decît formarea lui pe seama glicolizei sau a LR Creatinina se elimină cu urina

134
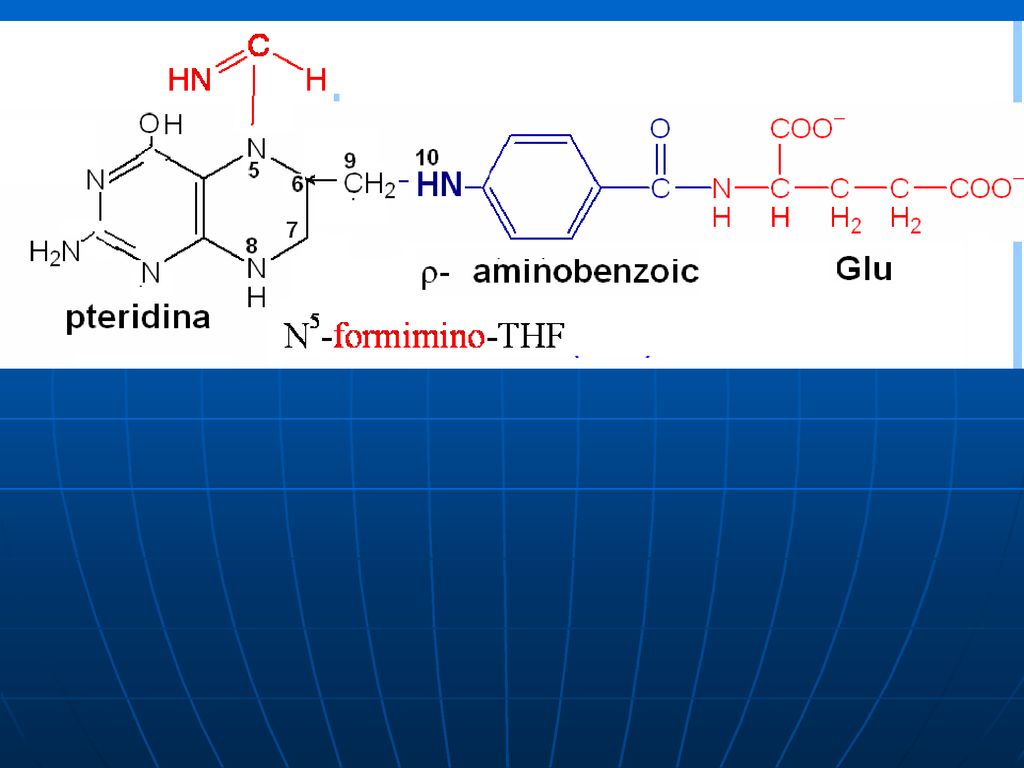
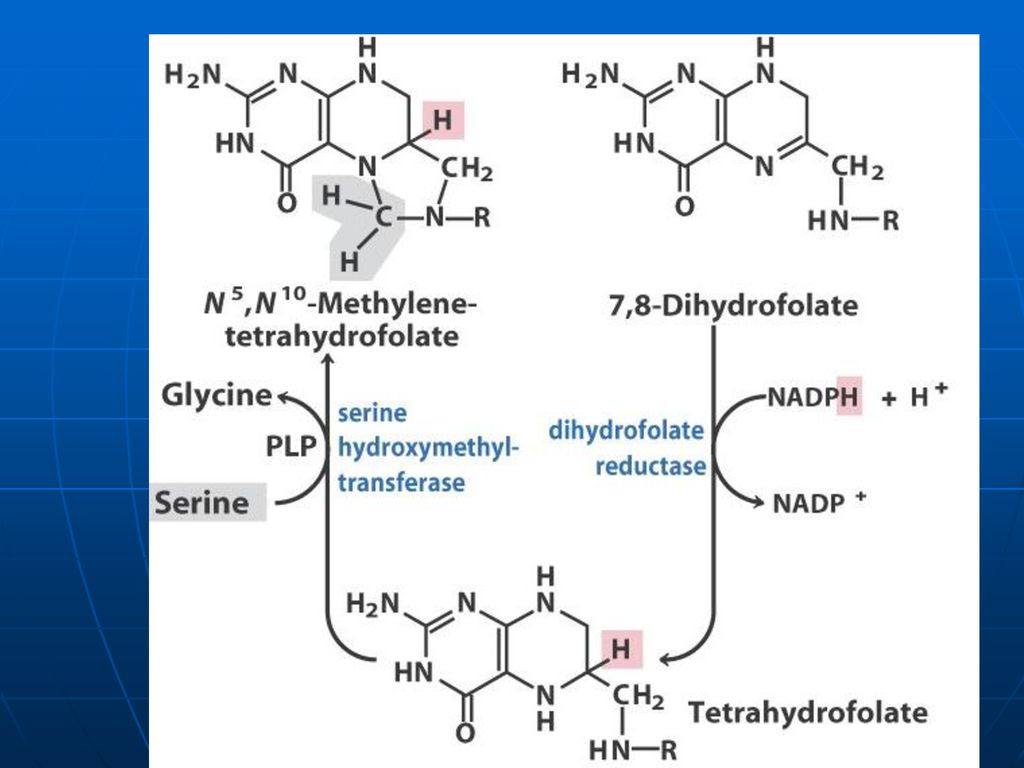
Acidul tetrahidrofolic - THF
Derivat al AF AF+NADPH+H------dihidrofolat +NADP DHF +NADPH+H-----THF +NADP

135
Rolul de transportator al unor fragmente cu un atom de carbon:
-metil (-CH3), -metilen (CH2-), -metenil (-CH=), -formil (- CH=O) -oximetil (-CH2-OH) -formil amino (-CH=NH) THF participă ca coenzimă în reacţiile de biosinteză a serinei, glicinei, metioninei, timinei.
, -metilen (CH2-), -metenil (-CH=), -formil (- CH=O) -oximetil (-CH2-OH) -formil amino (-CH=NH) THF participă ca coenzimă în reacţiile de biosinteză a serinei, glicinei, metioninei, timinei.")
136
La sinteza Gli (Ser)
")
138
Metabolismul glicinei, serinei şi cisteinei
Gli, Ser, Cis – AA neesenţiali, glucogeni A. Sinteza Gli: Tre Ser CO2+NH3+N5-N10-metilenTHF din etanolamina

139
A. Sinteza Ser: a. din Gli b. din 3-fosfoglicerat
c. din fosfatidilserina

140
Formation of Serine Glucose 3 Steps Inhibits Dehydrogenase Glycolysis
NAD+ NADH + H+ 3 Steps 3-Phospho- glycerate 3-Phospho- hydroxypyruvate Pyruvate Inhibits Glutamate Transaminase a-Ketoglutarate Phosphatase Serine (Ser) 3-Phosphoserine
 3-Phosphoserine.")
141
Sinteza Cis: din Ser +homocisteină din cistină

142
B. Catabolismul Gli: Gli ---Ser----Piruvat
Gli---a glioxilic (+NH3)----CO2+acid formic (captat de FH4) Gli---CO2+NH3+N5-N10-metilenTHF Ser:- piruvat: Serindehidratazei Prin transaminare cu piruvatul –hidroxipiruvat—2fosfoglicerat---fosfoenolpiruvat---piruvat Cis:- piruvat +sulfit (sulfat)
----CO2+acid formic (captat de FH4) Gli---CO2+NH3+N5-N10-metilenTHF. Ser:- piruvat: Serindehidratazei. Prin transaminare cu piruvatul –hidroxipiruvat—2fosfoglicerat---fosfoenolpiruvat---piruvat. Cis:- piruvat +sulfit (sulfat)")
144
Reacţiile metabolice a Gli
Gli participă la sinteza: Serinei Creatinei Hemului Glutationului AB conjugaţi Acidului hipuric purinelor Gli intră în componenţa colagenului Gli---–glicinamida ( intră în componenţa oxitocinei, vasopresinei)
")
146
Reacţiile metabolice a Ser
Ser participă la sinteza: Cis Gli Sfingolipidelor (SM) Etanolaminei (la sinteza cholinei) Fosfatidilserinei Fosfatidiletanolaminei
 Etanolaminei (la sinteza cholinei) Fosfatidilserinei. Fosfatidiletanolaminei.")
147
Reacţiile metabolice ale Cis
Formarea legăturilor disulfidice din proteine, E, Co La sinteza: glutationului taurinei (AB) fosfopanteteinei (grupare prostetică a PPA şi grupă funcţională a HSCoA)
 fosfopanteteinei (grupare prostetică a PPA şi grupă funcţională a HSCoA)")
148
AA dicarboxilici Asp şi Glu – AA neesenţiali, glucoformatori Sinteza:
prin reacţii de transaminare Din alfa cetoglutarat (Glu) Catabolismul: Asp - OA Glu – alfa - cetoglutarat
 Catabolismul: Asp - OA. Glu – alfa - cetoglutarat.")
150
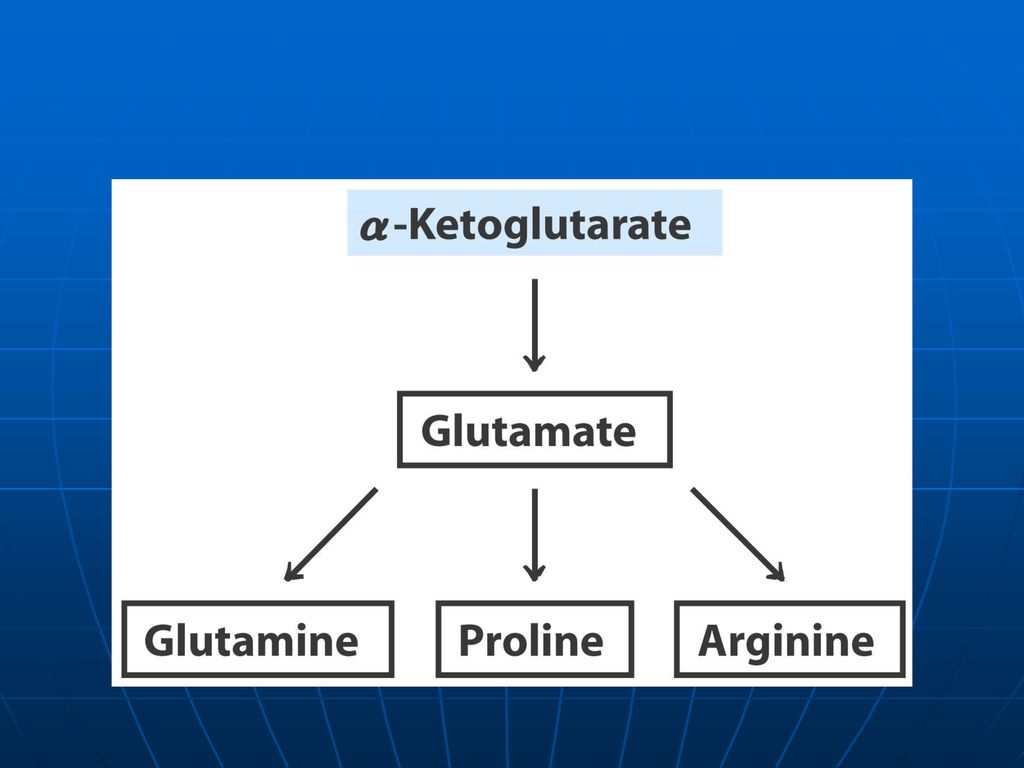
Reacţiile metabolice Glu participă la sinteza: Gln Pro Arg
γ aminobutiratului γ carboxiglutamatului Glutationului Este implicat în reacţiile de DO; transaminare

151
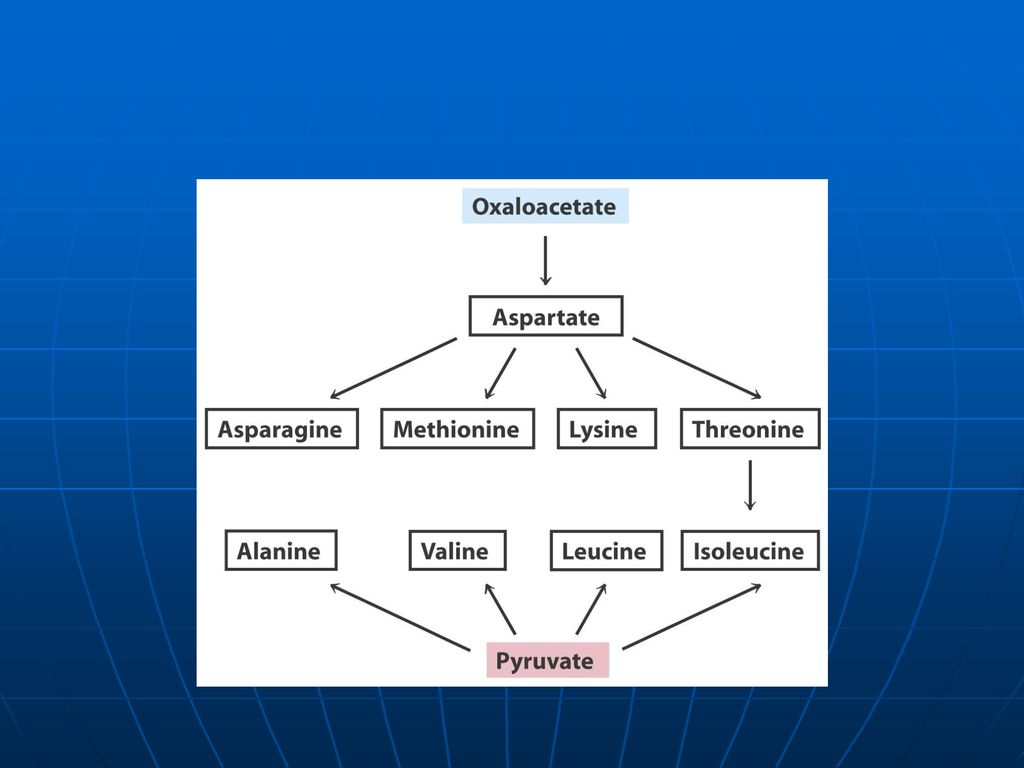
Reacţiile metabolice Asp participă la sinteza: Asn Ureei
BA purinice şi pirimidinice

154
METABOLISMUL NUCLEOPROTEINELOR

155
OBIECTIVELE Digestia şi absorbţia acizilor nucleici.
Biosinteza nucleotidelor purinice, reglarea. Biosinteza nucleotidelor pirimidinice, reglarea. Degradarea nucleotidelor purinice şi pirimidinice în ţesuturi. Guta.

156
Digestia şi absorbţia NP
NP alimentare se supun în TGI următoarelor modificări: în stomac - denaturarea NP - separarea componentei nucleinice de proteină. (P – se digeră după mecanismul clasic) În intestin – sub acţiunea endonucleazelor (dribonucleazelor sau ribonucleazelor, E pancreatice) – se scindează polinucleotidele pînă la oligonucleotide Sub acţiunea fosfodiesterazelor (pancreatice) – se eliberează 5-3 nucleotide, iar a nucleotidazelor (intestinale) are loc scindarea până la nucleozide şi P Nucleozidele sub acţiunea nucleozidazelor sunt scindate până la BA (purinice sau pirimidinice) şi pentoză (R sau dR)
 În intestin – sub acţiunea endonucleazelor (dribonucleazelor sau ribonucleazelor, E pancreatice) – se scindează polinucleotidele pînă la oligonucleotide. Sub acţiunea fosfodiesterazelor (pancreatice) – se eliberează 5-3 nucleotide, iar a nucleotidazelor (intestinale) are loc scindarea până la nucleozide şi P. Nucleozidele sub acţiunea nucleozidazelor sunt scindate până la BA (purinice sau pirimidinice) şi pentoză (R sau dR)")
157
Absorbţia R sau dR şi P – se absorb prin difuzie
BA purinice în celulele mucoasei intestinale sunt transformate în acid uric, eliminat apoi din circulaţie prin urină BA pirimidinice – se transportă cu ajutorul transportatorilor membranari O parte din produşii de digestie a NP – se absorb sub formă de nucleozide

158
BA purinice şi pirimidinice alimentare nu sunt utilizate la sinteza AN tisulari
Fondul nucleotidelor în organism se realizează prin: Sinteza de novo Conversia parţială a ribonucleotidelor în dribonucleotide Interconversia nucleotidelor Reutilizarea bazelor purinice

159
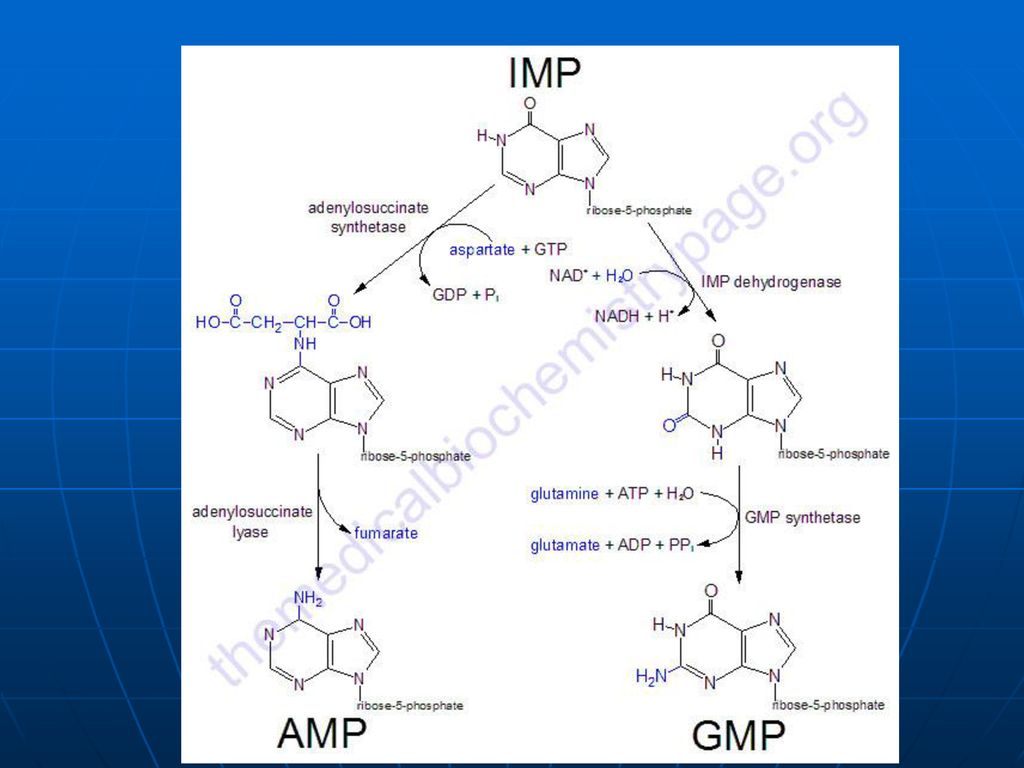
Biosinteza “de novo “a nucleotidelor purinice
1. Sinteza IMP Are loc în citozol Succesiune de 10 reacţii (Gli, Asp, Gln, CO2, FH4) Necesită Mg, K, ATP. Se consumă 6 legături ~P (proces exergonic) ireversibil Predomină în ficat
 Necesită Mg, K, ATP. Se consumă 6 legături ~P (proces exergonic) ireversibil. Predomină în ficat.")
161
2. Formarea 5-fosforibozil-aminei

162
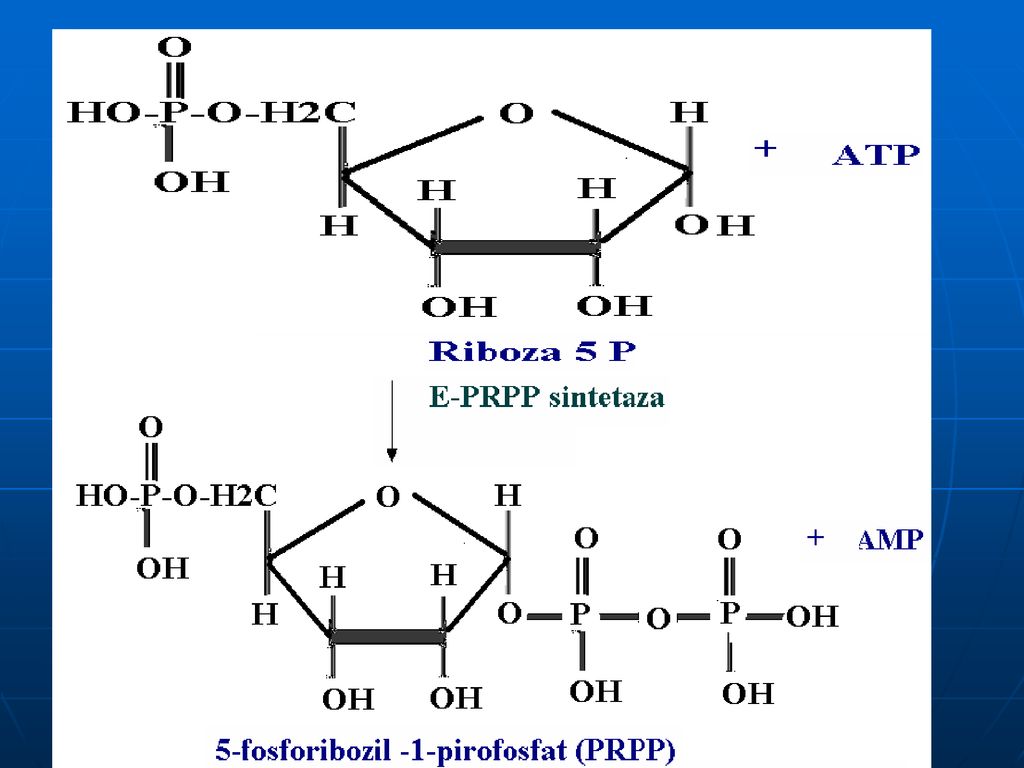
Reglarea La nivelul PRPP sintetazei: A: Pi
I: AMP, GMP, ATP,GTP,NAD, FAD, CoA Gln amidotransferaza: A: Gln, PRPP I: AMP,GMP,IMP, azaserina, acivicina (anologii structurali ai Gln)
")
163
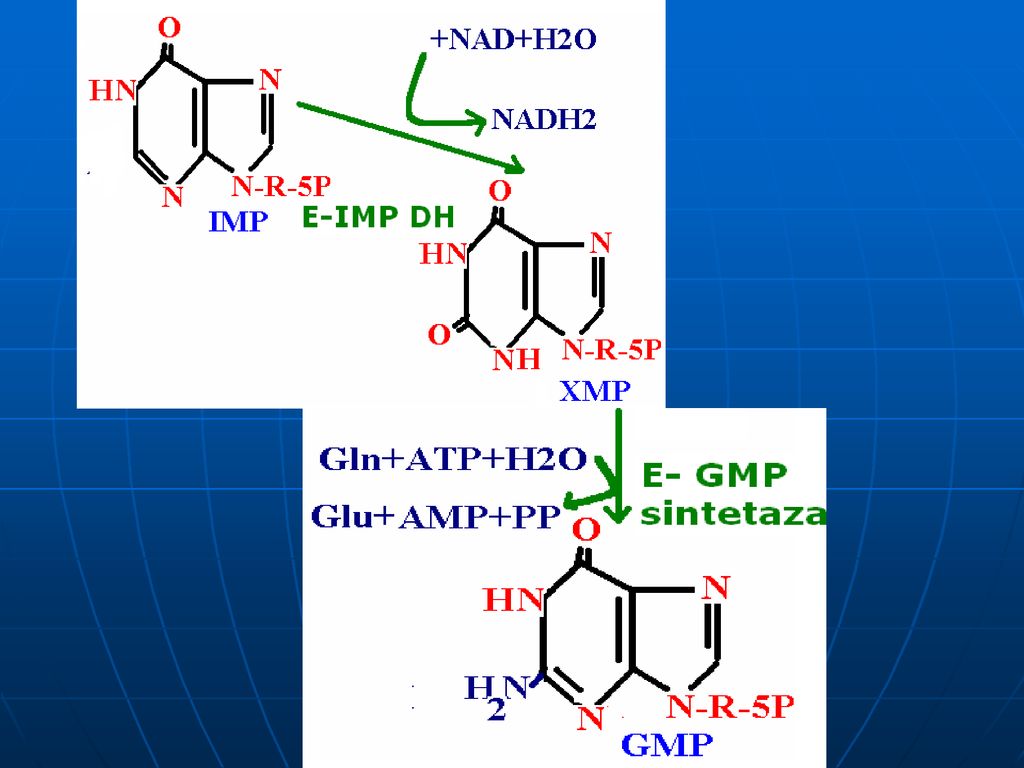
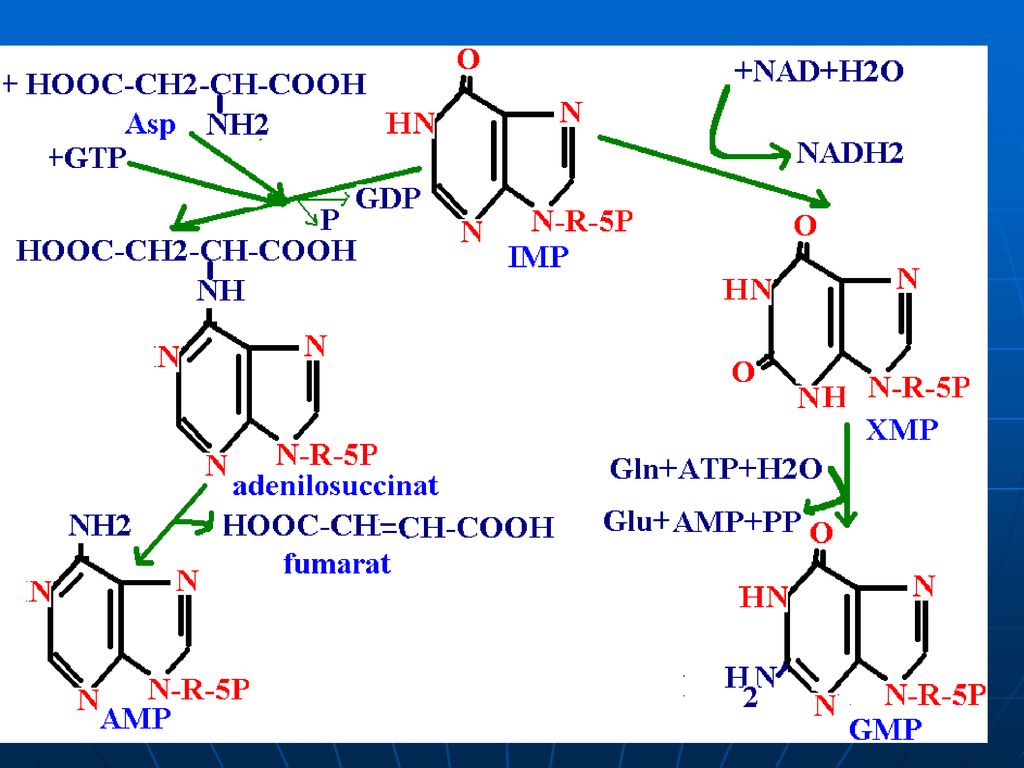
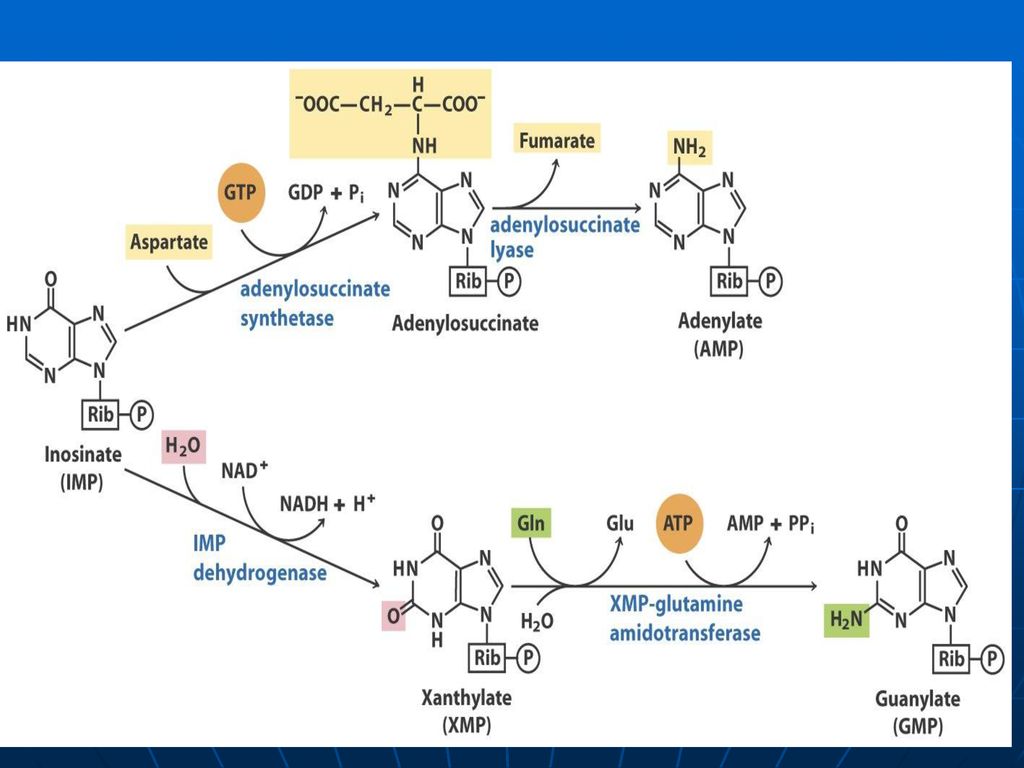
Sinteza inozin monofosfatului (IMP)
Glucoza-6-P Ciclul pentozofosfat PRPP sintetaza ATP AMP 5-fosforibozil 1 pirofosfat (PRPP) Riboza-5-P 5-fosfo--ribozilamină Glu PRPP amidotransferasa Gln inozin monofosfat (IMP) AA: Gli + Gln + Asp Cofactori: N10-formil THF N5N10-metenil THF Se consumă 6 leg P, Proces ireversibil Sinteza inozin monofosfatului (IMP)
 Riboza-5-P. 5-fosfo--ribozilamină. Glu. PRPP amidotransferasa. Gln. inozin monofosfat (IMP) AA: Gli + Gln + Asp. Cofactori: N10-formil THF. N5N10-metenil THF. Se consumă 6 leg P, Proces ireversibil. Sinteza inozin monofosfatului (IMP)")
164
Precursorii nucleului purinic

166
Sursa de atomi pentru IMP

172
Biosinteza nucleotidelor cu legături fosfat macroergice

173
Reglarea la nivelul AMP şi GMP
Inhibiţie feed-back de produşi finali: AMP-inhibă Adenilosuccinat sintetaza GMP- inhibă IMP DH Utilizarea “încrucişată” ca substrate ATP stimulează sinteza GMP GTP - stimulează sinteza AMP

174
Înhibiţia alosterică a sintezei purinelor;
PRPP sintetaza PRPP amido-transferaza IMP GMP AMP GDP ADP inhibiţia alosterică Înhibiţia alosterică a sintezei purinelor; ATP stimulează sinteza GMP; GTP stimulează sinteza AMP.

175
Interconversiunile şi reutilizarea purinelor
La hidroliza AN, nucleozidelor se formează BA purinice libere 1. Nucleotidele +H2O▬►nucleozid +Pi Pe acestă cale se formează inozina şi guanozina: IMP+H2O ▬►inozina +Pi GMP+H2O ▬►guanozina +Pi AMP+H2O ▬►adenozina +Pi Ultima reacţie este neînsemnată – adenozina se formează prin scindarea S-adenozil-homocisteinei

176
Nucleozidele sunt scindate la BA libere printr-o reacţie fosforolitică, sub acţiunea nucleozid fosforilazei: Nucleozid +Pi <▬► purina+ R-1-P IMP (GMP) +Pi <▬► hipoxantina (G) + R-1-P Adenina prin acestă cale nu se eliberează din adenozină. 2. dezaminarea (AMP-dezaminaza sau GMP-dezaminaza) AMP+H20 ▬► IMP+NH3 GMP+H20 ▬► xantină +NH3
 +Pi <▬► hipoxantina (G) + R-1-P. Adenina prin acestă cale nu se eliberează din adenozină. 2. dezaminarea (AMP-dezaminaza sau GMP-dezaminaza) AMP+H20 ▬► IMP+NH3. GMP+H20 ▬► xantină +NH3.")
177
Aceste împreună cu BA sintetizate de novo alcătuiesc fondul metabolic comun accesibil tuturor celulelor

178
Reutilizarea bazelor purinice
Purinele libere se reutilizează în nucleotide şi sunt utilizate din nou la sinteza AN. Se cunosc 2 căi de reîncorporare a bazelor purinice în nucleozide (sau nucleotide) I. Condensarea BA cu PRPP 1. Adenina + PRPP ---AMP +PP E – adenilofosforiboziltransferaza 2. Guanina +PRPP –GMP +PP 3. Hipoxantina +PRPP – --- IMP +PP E – hipoxantin guanilatfosforiboziltransferaza Sinteza din produse finite este mai economă pentru celule decît sinteza de novo.
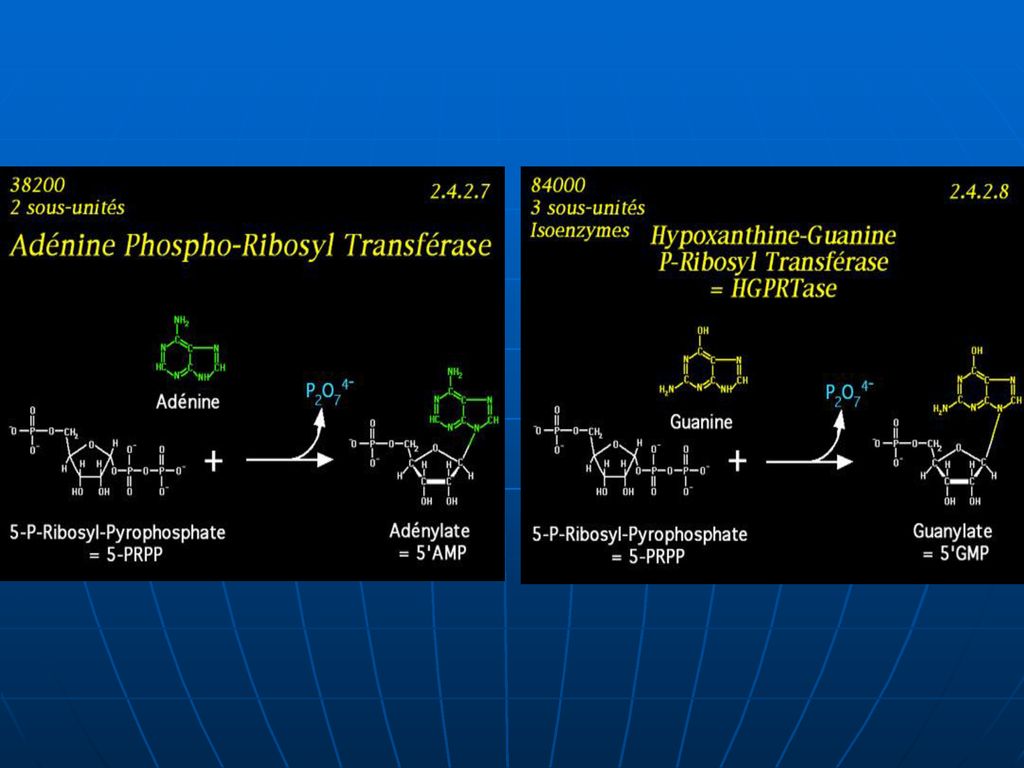
 I. Condensarea BA cu PRPP. 1. Adenina + PRPP ---AMP +PP. E – adenilofosforiboziltransferaza. 2. Guanina +PRPP –GMP +PP. 3. Hipoxantina +PRPP – --- IMP +PP. E – hipoxantin guanilatfosforiboziltransferaza. Sinteza din produse finite este mai economă pentru celule decît sinteza de novo.")
180
II cale: Încorporarea purinei în nucleotid în două etape (minoră):
2a.ribozo-1-fosfat + purină <▬► nucleozid+H3PO4 E-nucleozidfosforilaza 2b. nucleozid + ATP ▬► nucleotid +ADP E- nucleozidkinaza

181
Biosinteza dezoxiribonucleotidelor
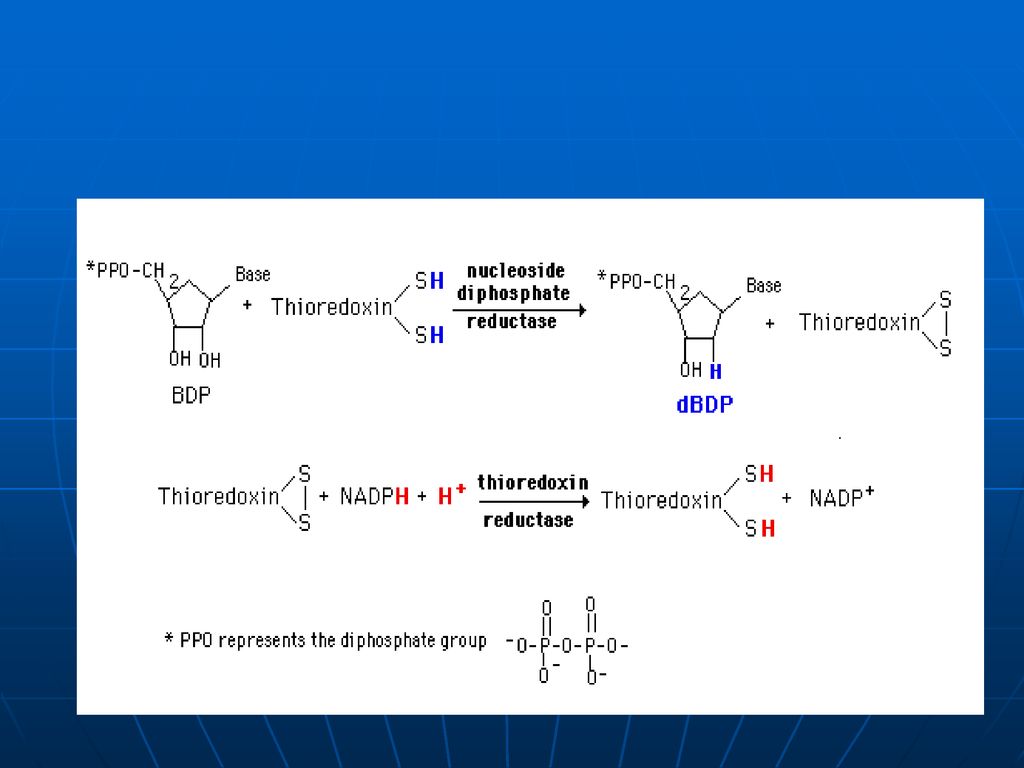
Reducerea de ribozil (în poziţia 2) din nucleozid difosfaţi în 2- dezoxiribozil 1. Echivalenţii reducători de pe NADPH+H sunt transferaţi pe o proteină mică – tioredoxina , 2. sub acţiunea tioredoxin reductazei – tioredoxina se reduce. 3. sub acţiunea ribonucleozid reductazei se reduce restul ribozil la dezoxiribozil
 din nucleozid difosfaţi în 2- dezoxiribozil. 1. Echivalenţii reducători de pe NADPH+H sunt transferaţi pe o proteină mică – tioredoxina , 2. sub acţiunea tioredoxin reductazei – tioredoxina se reduce. 3. sub acţiunea ribonucleozid reductazei se reduce restul ribozil la dezoxiribozil.")
184
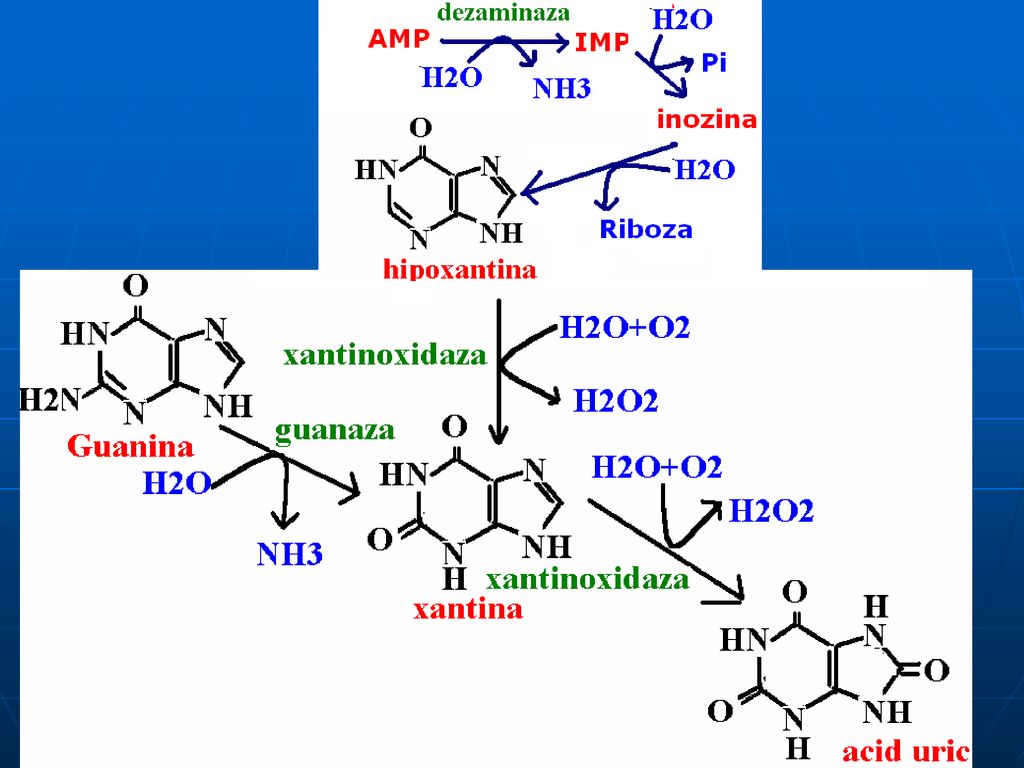
Catabolismul purinelor

186
Acidul uric Acidul uric se formează din: Nucleotide exogene (intestin)
Din AMP şi GMP rezultate din degradarea AN tisulari Din GMP şi AMP sintetizaţi de novo Acidul uric – compus greu solubil în H2O. În plasmă şi lichidele intersteţiale se găseşte ca sare monosodică- monourat de sodiu, fiind ceva mai solubil. Este un AO puternic Excreţia de acid uric în 24 ore este de mg.

187
Guta Se caracterizează prin hiperuricemie. Deosebim:
Primară – rezultat al erorilor înăscute a metabolismului Secundară – cauzată de alte maladii (cancer, insuficienţă renală cronică, traumatisme, chimioterapii, infecţii cronice, acidoza metabolică)
")
188
Guta Dureri artritice episodice, cronice – reacţia inflamatorie declanşată de cristalele de urat fagocitate de leucocite Nefrolitiază – favorizată formarea calculilor de urat (în urinele mai acide şi de acid uric) Depozite de acid uric în ţesuturi moi (tofi gutoşi) – creşterea c% uratului în sânge, depăşirea pragului de solubilitate, determină precipitarea uratului monosodic în jurul articulaţiilor de la extremităţi
 Depozite de acid uric în ţesuturi moi (tofi gutoşi) – creşterea c% uratului în sânge, depăşirea pragului de solubilitate, determină precipitarea uratului monosodic în jurul articulaţiilor de la extremităţi.")
189
Guta
190
Guta. Etiopatogeneza. Factorul decesiv al hiperuricemiei – este creşterea c% de PRPP, rezultatul unei sinteze crescute sau încetinirii ritmului de utilizare. Deficitele enzimatice ce măresc nivelul de PRPP (acid uric) pot fi: PRPP-sintetaza – activitate catalitică crescută (sensibilitate redusă la I) Deficienţa de hipoxantin-guanin- fosforibozil transferazei (HGPRT) -reutilizarea guaninei şi hipoxantinei – la sinteza de IMP şi GMP Deficit de Gl-6 fosfotază (Gl 6 P nu ia calea gluconeogenezei dar a ciclului pentozofosfat – creşte c% de R-5P – creşte C% şi de PRPP.
 pot fi: PRPP-sintetaza – activitate catalitică crescută (sensibilitate redusă la I) Deficienţa de hipoxantin-guanin- fosforibozil transferazei (HGPRT) -reutilizarea guaninei şi hipoxantinei – la sinteza de IMP şi GMP. Deficit de Gl-6 fosfotază (Gl 6 P nu ia calea gluconeogenezei dar a ciclului pentozofosfat – creşte c% de R-5P – creşte C% şi de PRPP.")
191
Tratamentul gutei Administrare de alopurinol (analog structural al hipoxantinei) – inhibă xantinoxidaza şi împedică transformarea hipoxantinei în xantină şi în acid uric. Hipoxantina şi xantina (sunt mai solubile( nu se depun în ţesuturi şi sunt excretate ca produşi finali ai purinelor.
 – inhibă xantinoxidaza şi împedică transformarea hipoxantinei în xantină şi în acid uric. Hipoxantina şi xantina (sunt mai solubile( nu se depun în ţesuturi şi sunt excretate ca produşi finali ai purinelor.")
192
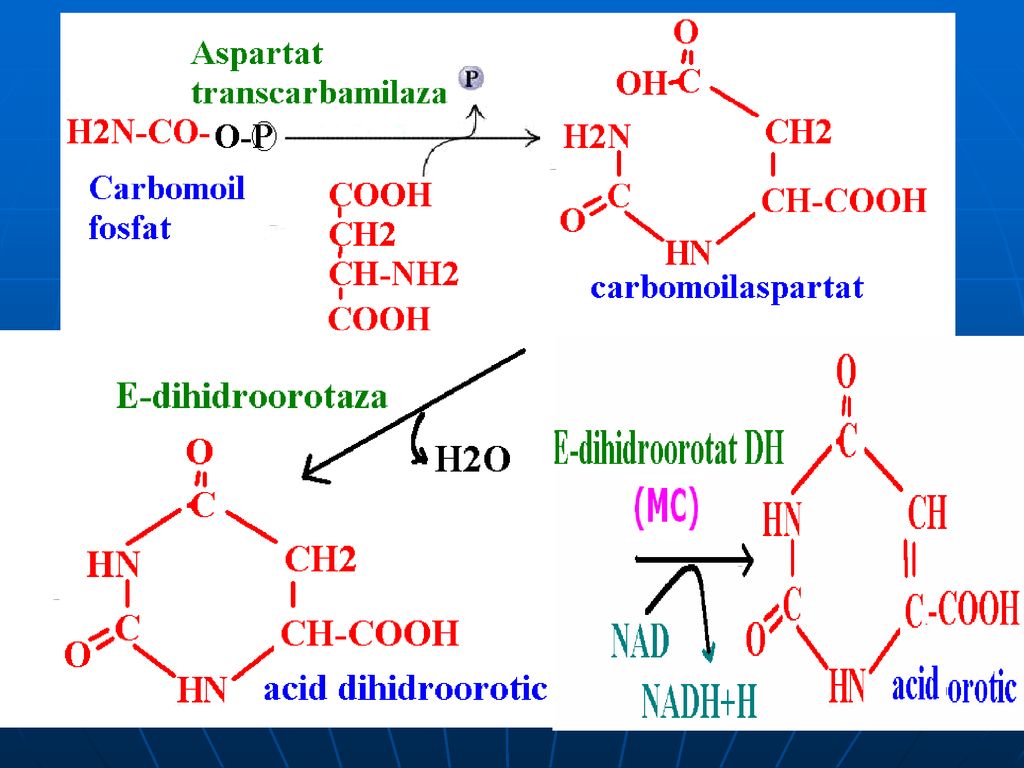
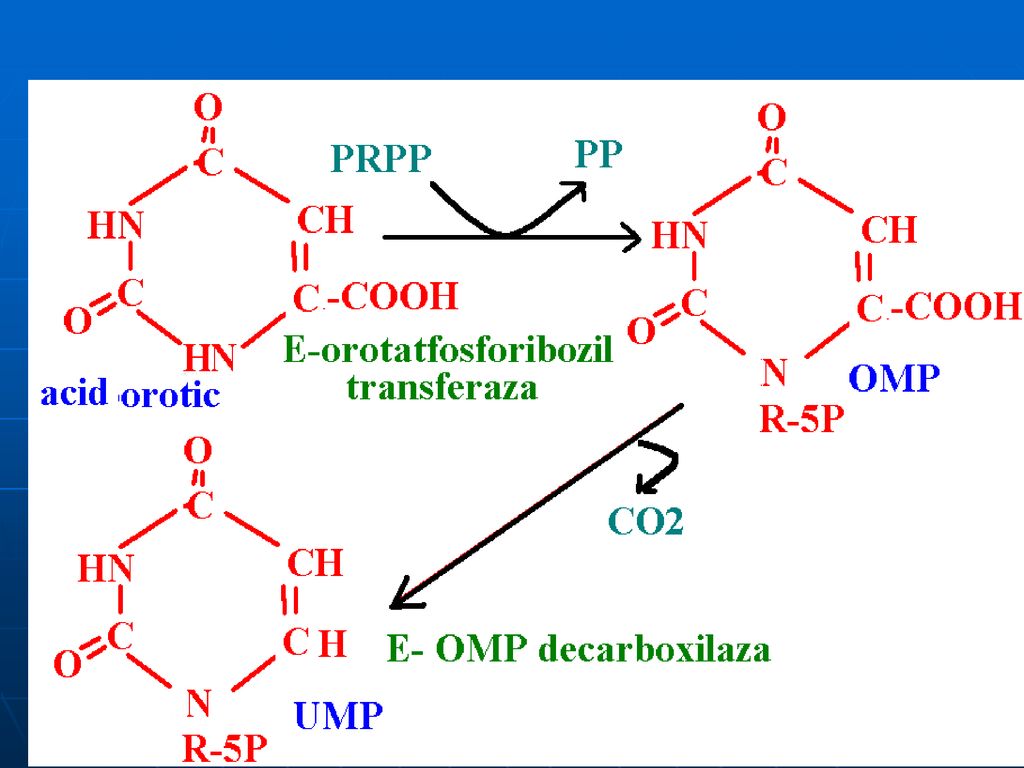
Biosinteza de novo a nucleotidelor pirimidinice
Precursorii nucleului pirimidinic:

193
Biosinteza de novo a nucleotidelor pirimidinice
1. Formarea carbomoil fosfatului (citozolică)
")
196
Formarea de UTP şi de CTP
1. UMP +ATP ▬►UDP+ADP 2. UDP +ATP ▬►UTP+ADP CTP se formează din UTP sub acţiunea CTP- sintetazei:

197
Sinteza CTP

198
Sinteza de d-TMP Se formează din dUMP dUDPdUTPdUMP dTMP
dCDPdCMPdUMP

200
INHIBITORS OF N5,N10 METHYLENETETRAHYDROFOLATE REGENERATION
dUMP dTMP thymidylate synthase DHF N5,N10 – METHYLENE-THF X NADPH + H+ GLYCINE FdUMP dihydrofolate reductase serine hydroxymethyl transferase NADP+ SERINE X THF METHOTREXATE AMINOPTERIN TRIMETHOPRIM

201
Reglarea metabolismului pirimidinic
dATP – inhibă reducerea sa şi stimulează reducerea dUDP şi dCTP TTP – inhibă reducerea pirimidinelor şi stimulează reducerea purinelor.

202
Reutilizarea nucleotidelor pirimidinice
BA pirimidinice nu sunt reutilizate ci degradate (beta-Ala, beta-aminoizobutiric +CO2 +NH3)
")
203
Catabolismul pirimidinelor

204
Catabolismul pirimidinelor

205
Catabolismul pirimidinelor

206
METABOLISMUL CROMOPROTEINELOR

207
Obiectivele Structura chimică şi rolul biologic al cromoproteinelor.
Digestia şi absorbţia cromoproteinelor. Biosinteza hemului. Reglarea procesului. Catabolismul hemoglobinei în ţesuturi. Legătura dintre pigmenţii sanguini, biliari, urinari şi a maselor fecale. Importanţa determinării lor în diagnosticul şi diferenţierea icterelor. Metabolismul fierului în organism.

208
Structura chimică şi rolul biologic al CP
proteine conjugate: partea proteică+ partea neproteică: pigment (substanţă colorată). Reprezentanţii: clorofila, hemoproteidele Flavoproteidele
. Reprezentanţii: clorofila, hemoproteidele. Flavoproteidele.")
209
Rolul: participă în fotosinteză,
respiraţia tisulară, reacţiile de oxido-reducere transportul oxigenului şi CO2 senzaţiile de lumină şi culoare

210
Hemoproteidele substanţe complexe alcătuite din proteine + hem (heterocicluri tetrapirolice neproteice) şi ioni ai metalelor Reprezentanţii principali: hemoglobina mioglobina, citocromii, catalaza peroxidaza

212
Structura hemului 4 inele pirolice +Fe +punţi metinice (α, β, γ, δ)
4 radicali metil 2 vinil 2 resturi de a propionic

213
Digestia hemoproteinelor
în tractul digestiv sub influenţa E - se scindează în componenta proteică şi hem. Proteina simplă degradează până la AA după mecanismul clasic Hemul - nu se supune transformărilor şi este eliminat cu masele fecale.

214
Biosinteza Hemului Succinil CoA + Glicina Ferrohelataza HEM Fe2+ -Aminolevulinat sintaza piridoxal fosfat dependentă -Aminolevulinat Coproporfirinogen III Protoporfirina IX MITOCONDRIA -Aminolevulinat (două molecule) CITOPLASMA Aminolevulinat dehidrataza Enzima conţine zinc Porfobilinogen 4 molecule combinate Uroporfirinogen III Coproporfirinogen III
 CITOPLASMA. Aminolevulinat dehidrataza. Enzima conţine zinc. Porfobilinogen. 4 molecule. combinate. Uroporfirinogen III. Coproporfirinogen III.")
215
Biosinteza hemului Substanţele iniţiale în sinteza hemului sunt Gli şi succinil-CoA, Localizare: în toate ţesuturile, dar cu intensitate mai mare în celulele sistemului eritroformator din măduvă, ficat şi splină. Etapele: sinteza acidului aminolevulinic Formarea porfobilinogenului Formarea protoporfirinei IX Unirea protoporfirinei IX cu Fe2+

216
E- aminolevulinatsintaza (mitocondrială)
")
217
ALS este o enzimă: mitocondrială piridoxal fosfat şi Mg++ dependentă
masa moleculară de D. Reglarea -ALA sintazei (alosterică): este inhibată de hem se reglează prin inducţie-represie (sinteza este indusă prin scăderea c% hemului; iar represia – invers) Acţiune inductoare o au: barbituricele, insecticidele, sulfamidele, h. estrogeni Acţiune represoare: glucoza Hipoxia – măreşte activitatea E în ţesuturile eritropoietice, fără efect în ficat
: este inhibată de hem. se reglează prin inducţie-represie (sinteza este indusă prin scăderea c% hemului; iar represia – invers) Acţiune inductoare o au: barbituricele, insecticidele, sulfamidele, h. estrogeni. Acţiune represoare: glucoza. Hipoxia – măreşte activitatea E în ţesuturile eritropoietice, fără efect în ficat.")
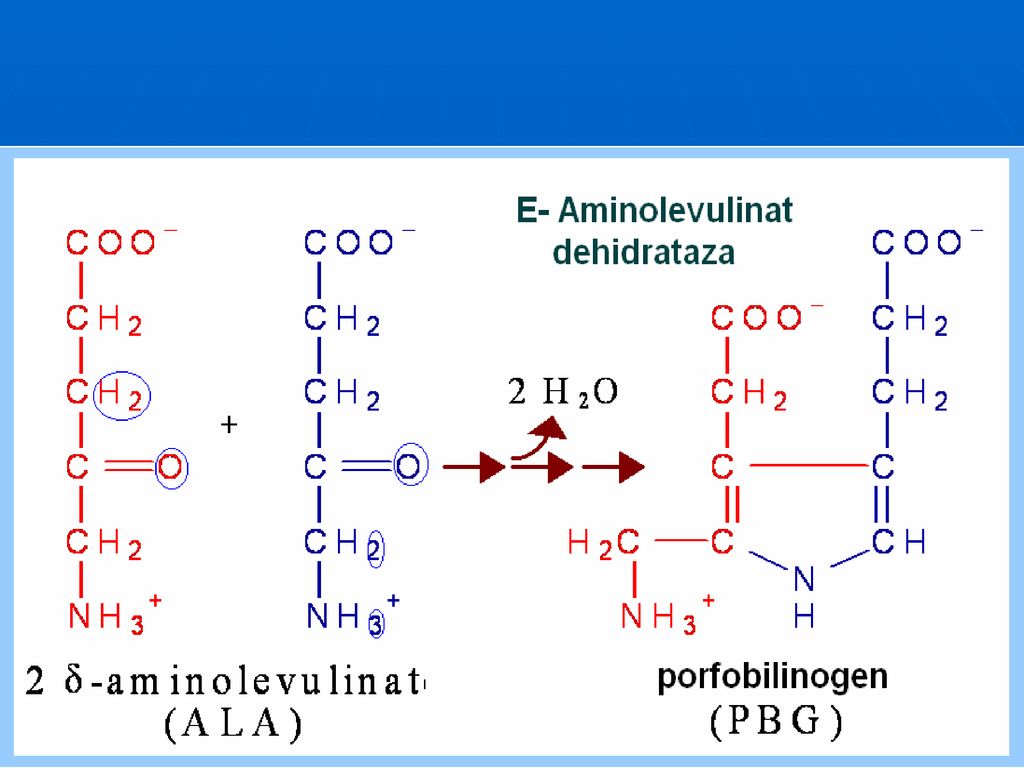
219
Aminolevulinat dehidrataza
este o E citoplasmatică, are ca cofactor ionul de Zn şi PALP. Este inhibată alosteric de hem şi hemoproteine. Activitatea sa este diminuată în saturnism (intoxicaţie cu Plumb) şi în alcoolism (acut sau cronic).
 şi în alcoolism (acut sau cronic).")
220
Porphyrin from δ-aminolevulinate/heme (ferrochelatase)
")
222
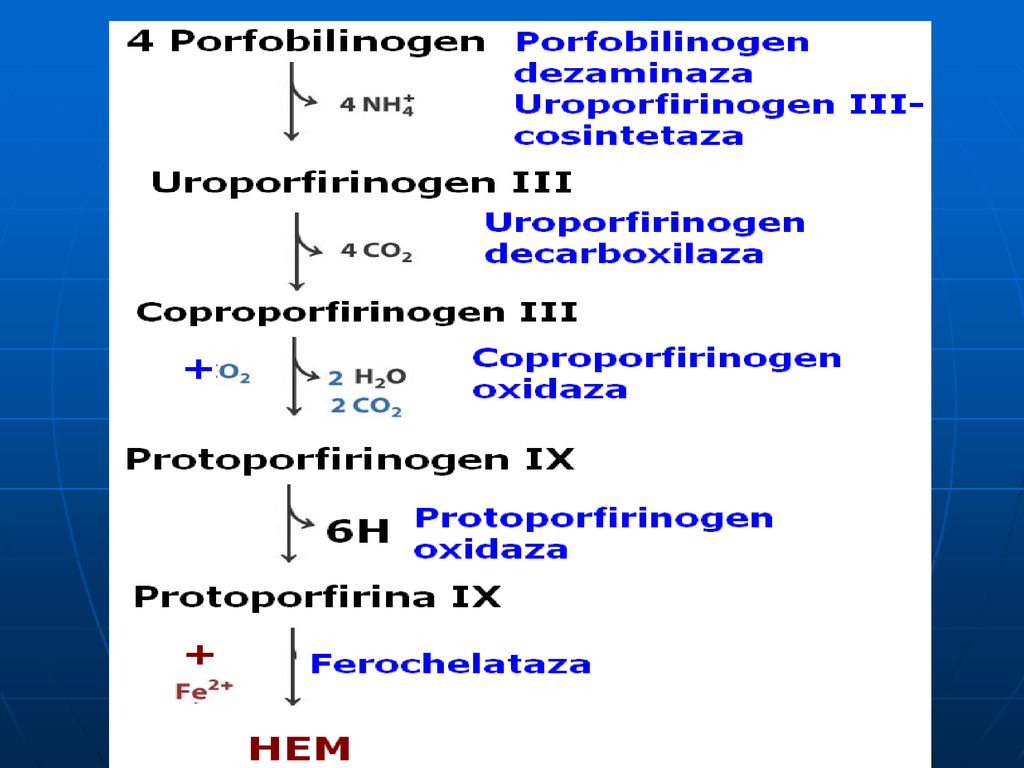
Patru molecule de porfobilinogenă se condensează cu formare de uroporfirinogen III
este prezentă în citoplasma hepatocitelor. E- porfobilinogendezaminaza+uroporfirinogencosintaza este termolabilă (se denaturează la 60°C).
.")
223
Decarboxilarea uroporfirinogenului III
Uroporfirinogen decarboxilaza - este o E citoplasmatică (4 radicali de acid acetic – metil).
.")
224
Oxidarea Coproporfirinogenului III
Coproporfirinogen oxidaza: - E mitocondrială (decarboxilează şi dehidrogenează oxidativ) ce transformă doi radicali propionil în vinil.
 ce transformă doi radicali propionil în vinil.")
225
Oxidarea protoporfirinogenului
Protoporfirinogen oxidaza catalizează formarea legăturilor duble în inelul porfirin

226
Adiţionarea Fe Ferochelataza fixează atomul de Fer cu formare de hem.
Există mai multe izoenzime a ferochelatazei în mitocondrii sau în citoplazmă care conduc la sinteza de hemoglobină, citochromi.

227
Porfiriile boli metabolice produse de defectele enzimatice în procesul de biosinteză a hemului Se caracterizează prin supraproducţia, acumularea şi eliminarea precursorilor de hem

228
Clasificarea porfiriilor
1. primare – cauzate de defecte enzimatice ereditare 2. Secundare – sunt consecutive altor afecţiuni ( diabet, intoxicaţie) Porfiriile primare după localizare pot fi: - eritropoietice - hepatice - mixte
 Porfiriile primare după localizare pot fi: - eritropoietice. - hepatice. - mixte.")
229
Porfiriile eritropoietice
Porfiria eritropoietică congenitală (Gunther) Protoporfiria
 Protoporfiria.")
230
Porfiriile hepatice Porfiria acută intermitentă Porfiria variegata
Coproporfiria ereditară Porfiria cutanea tarda

231
PORFIRIILE Agent Orange Porfiria deficienţei ALA-dehidratazei
Mitochondria GLICINa + SuccinilCoA d-aminolevulinat(ALA) Porfobilinogen(PBG) hidroximetilbilan uroporfirinogen III coproporfirinogen III Protoporfirinogen IX protoporfrin IX Hem Agent Orange ALA sintaza 3p21/Xp11.21 Porfiria deficienţei ALA-dehidratazei ALA dehidrataza 9q34 Porfiria Acută intermitentă PBG dezaminaza 11q23 Uroporfirinogen III cosintaza Porfiria eritropoietică congenitală 10q26 Uroporfirinogen decarboxilaza Porfiria cutanea tarda 1q34 Coproporfirinogen oxidaza coproporfiria erediatară 9 Protoporfirinogen oxidaza porfiria Variegată 1q14 Ferrohelataza protoporfiria eritropoietică 18q21.3
 Porfobilinogen(PBG) hidroximetilbilan. uroporfirinogen III. coproporfirinogen III. Protoporfirinogen IX. protoporfrin IX. Hem. Agent Orange. ALA sintaza. 3p21/Xp Porfiria deficienţei. ALA-dehidratazei. ALA dehidrataza. 9q34. Porfiria Acută. intermitentă. PBG dezaminaza. 11q23. Uroporfirinogen III. cosintaza. Porfiria eritropoietică. congenitală. 10q26. Uroporfirinogen. decarboxilaza. Porfiria. cutanea tarda. 1q34. Coproporfirinogen. oxidaza. coproporfiria. erediatară. 9. Protoporfirinogen. oxidaza. porfiria. Variegată. 1q14. Ferrohelataza. protoporfiria. eritropoietică. 18q21.3.")
232
Porfiria eritropoietică congenitală
- afecţiune rară - autosomal recesivă Cauza: sinteza defectuoasă a uroporfirinogen III cosintetazei Supraproducerea de uroporfirinogen I şi coproporfirinogen I (elimină prin urină şi masele fecale) – urina e de culoare roşie Eritrocitele se distrug prematur Clinic: Hepatomegalie Fotosensibilitate mare cu producerea de eriteme şi vezicule ce lasă cicatrice Dinţii roşii Anemie hemolitică “Setea de sânge”
 – urina e de culoare roşie. Eritrocitele se distrug prematur. Clinic: Hepatomegalie. Fotosensibilitate mare cu producerea de eriteme şi vezicule ce lasă cicatrice. Dinţii roşii. Anemie hemolitică. Setea de sânge")
233
Protoporfiria este determinată de deficienţa sintezei ferochelatazei
Eritrocitele, plasma şi masele fecale conţin în cantităţi mari protoporfirina IX Reticulocitele şi pielea prezintă fluorescenţă roşie Ciroză urticărie

234
Porfiria acută intermitentă
Activitatea scăzută a uroporfirinogensintetazei Creştereas c% de aminolevulinat şi porfobilinogen (se elimină cu urina, ei sunt incolori, dar în contact cu aerul şi lumina se polimerizează – închid culoarea urinei) Simptome: Dureri abdominale Paralizii periferice Tulburări ale SNC
 Simptome: Dureri abdominale. Paralizii periferice. Tulburări ale SNC.")
235
Porfiria cutanea tarda
Cea mai frecventă E cauzată de deficitul uroporfirinogen decarboxilaza se măreşte c% uroporfirinogen I şi III Manifestările clinice: Fotosensibilitatea cutanată (eriteme, vezicule, cicatrice Tulburări abdominale Tulburări neurologice Fluorescenţa ficatului

236
Coproporfiria ereditară
Defect enzimatic în sinteza coproporfirinogenoxidaza (mitocondrială) eliminarea renală şi prin masele fecale a unor cantităţi excesive de coproporfirinogen III (în contact cu aerul se oxidează la coproporfirină III, care este colorată în roşu) Clinic: simptomele porfiriei acute intermitente + fotosensibilitatea cutanată
 eliminarea renală şi prin masele fecale a unor cantităţi excesive de coproporfirinogen III (în contact cu aerul se oxidează la coproporfirină III, care este colorată în roşu) Clinic: simptomele porfiriei acute intermitente + fotosensibilitatea cutanată.")
237
Porfiria variegata Micşorarea sintezei protoporfirinogen oxidazei + ferochelatazei Mărirea c% de protoporfirină, coproporfirină, uroporfirină La debutul bolii – se măreşte aminolevulinatul şi porfobilinogenul în urină Apare o porfirină atipică- X – hidrofilă, ce are ataşat un rest peptidilic Simptomele clinice – ca la coproporfiria eriditară

238
Catabolismul Hb Zilnic se degradează 6 g Hb (300 mg de hem) Durata vieţii eritrocitelor este de 120 zile Ruperea membranelor celulelor îmbătrînite ale eritrocitelor – eliberarea Hb 1. Hb + haptoglobina – se formează complexul Hb-Haptoglobină, fagocitat de macrofagele sistemului RE (reticulului endotelial) în special al ficatului, splinei şi ganglionilor limfatici
 în special al ficatului, splinei şi ganglionilor limfatici.")
239
în RE al ficatului, splinei şi ganglionilor limfatici
2.a. oxidarea microsomială a complexului sub acţiunea hemoxigenazei microsomiale - se obţine un intermediar – hidroxihemina (gr OH la “C” metinic şi fierul în stare oxidată (Fe3+). 2b. scindarea punţii metinice sub acţiunea hemoxigenazei (monooxigenază solicitantă de O2 şi NADPH, se elimină CO – verdoglobina
. 2b. scindarea punţii metinice sub acţiunea hemoxigenazei (monooxigenază solicitantă de O2 şi NADPH, se elimină CO – verdoglobina.")
240
3. Verdoglobina – pierde Fe şi globina – se transformă în biliverdină
3. Verdoglobina – pierde Fe şi globina – se transformă în biliverdină. Globina este hidrolizată la AA, iar Fe se leagă de transferină (este reciclat sau depozitat în ficat) Biliverdina – pigment biliar de culoare verde.
 Biliverdina – pigment biliar de culoare verde.")
241
4. reducerea biliverdinei (NADPH+H) la nivelul punţii γ metinice /biliverdinreductazei/ - bilirubina (galben-portocalie)
 la nivelul punţii γ metinice /biliverdinreductazei/ - bilirubina (galben-portocalie).")
242
5. În sînge: bilirubina se leagă cu albumina şi este transportată la ficat
Bilirubină liberă indirectă – 75% din toată cantitatea (2,5-10 mg/l; 8,7-17 µmol/L) Este toxică Nu trece prin filtrul renal Nu se elimină prin bilă Reacţie indirectă cu diazoreactivul
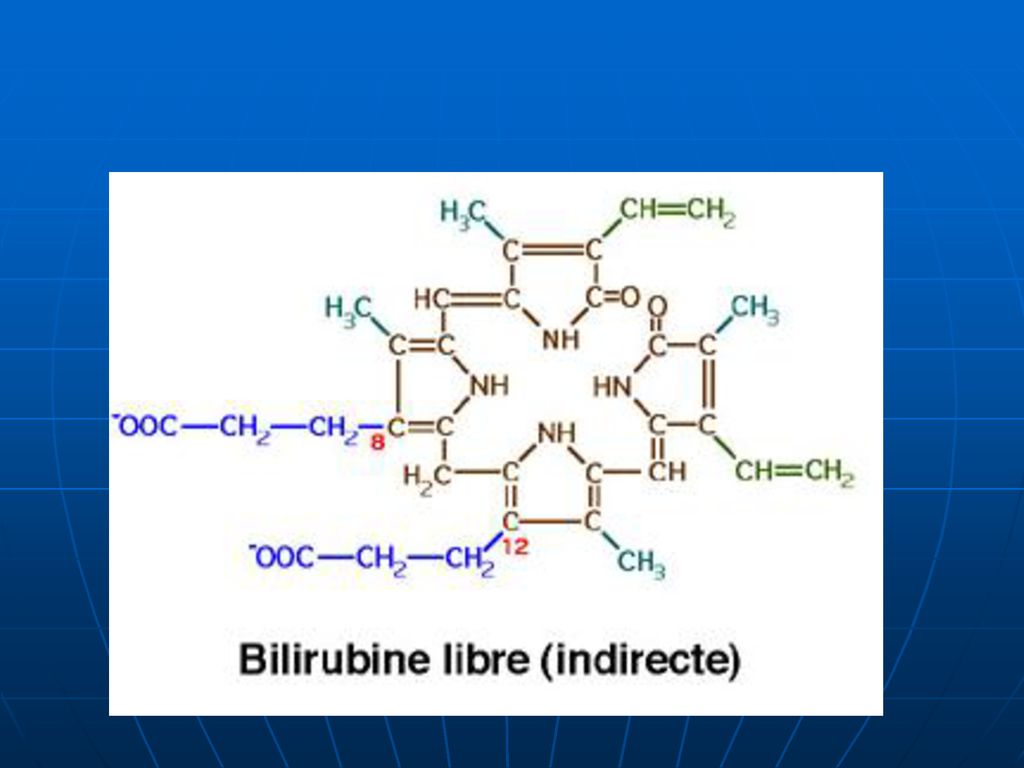
 Este toxică. Nu trece prin filtrul renal. Nu se elimină prin bilă. Reacţie indirectă cu diazoreactivul.")
244
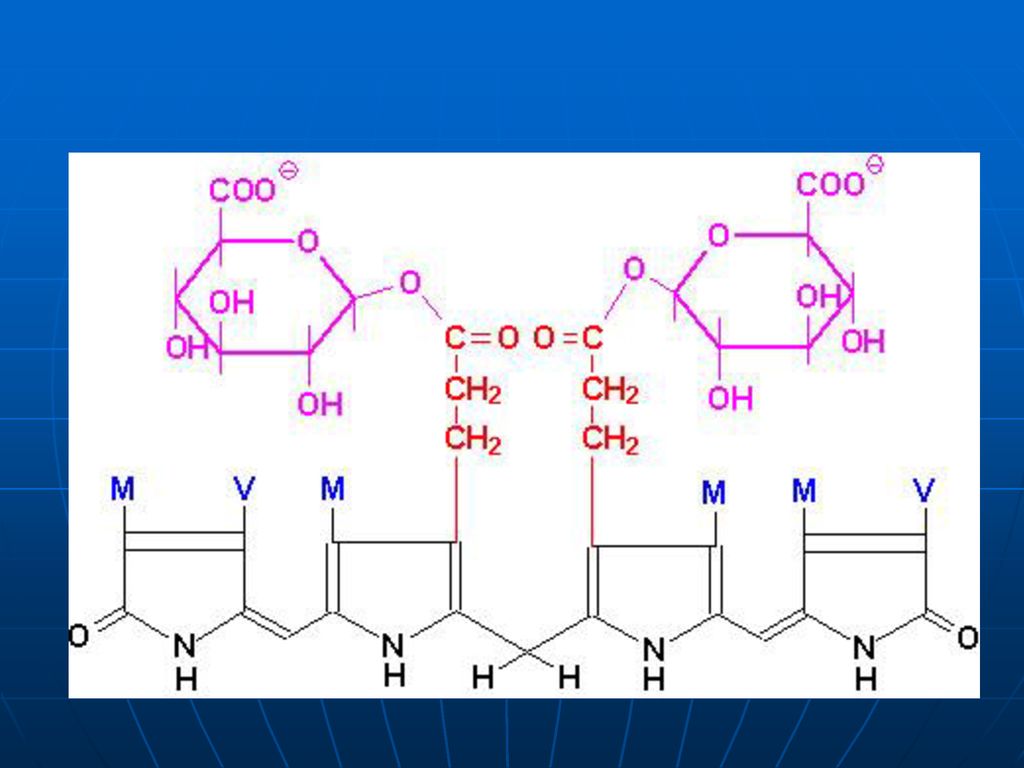
6. În ficat: sub acţiunea bilirubin-UDP-glucoronil-transferazei, bilirubina se conjugă cu a glucuronic activat (UDP glucuronat) – mono şi diglucuronid (hidrosolubili) Bilirubina conjugată, directă Valoarea medie: 2,6 µM/L Sub formă de glucuronid se excretă prin bilă – în intestinul subţire (o cantitate f mică reabsorbită – ficat), dar cea mai mare parte trece în intestinul gros
, dar cea mai mare parte trece în intestinul gros.")
246
7. În ileonul terminal şi intestinul gros glucuronidaza (produsă de bacteriile microflorei intestinale) înlătură resturile acidului glucuronic şi transformă bilirubina în mesobilirubină, care suferă o serie de reduceri mesobilinogen (urobilinogen), care prin reduceri ulterioare se va transforma în stercobilinogen. Ultimul cu masele fecale se elimină în mediul ambiant – se oxidează pînă la stercobilină

247
8. O parte din mesobilinogen (urobilinogen), se reabsoarbe în sistemul v portae – se intorc în ficat – se scindează la dipiroli, care se elimină cu bila în intestin – ciclul entero-hepatic al pigmenţilor biliari 9. alta - se reabsoarbe la nivelul venelor hemoroidale, care prin sistemul v cava inferior nimereşte în sistemul general. Se elimină pe cale renală cu urina (0,5-2,4 mg timp de 24 ore)
")
248
Catabolismul hemoglobinei
Eritrocit Stercobilin excreted in feces Urobilinogen formed by bacteria Excreţia cu urina Urobilin Heme Globin Hemoglobina RINICHI CO Biliverdina IX Heme oxygenase O2 Reabsorbţia in sînge INTESTIN Cu bila spre intestin Bilirubina (insolubilă în apă) NADP+ NADPH Biliverdin reductaza Bilirubin diglucuronat (solubil în apă) 2 UDP-glucuronat Ficat Bilirubin (insolubilă în apă) Prin sînge spre ficat Catabolismul hemoglobinei
 NADP+ NADPH. Biliverdin. reductaza. Bilirubin diglucuronat. (solubil în apă) 2 UDP-glucuronat. Ficat. Bilirubin. (insolubilă în apă) Prin sînge spre ficat. Catabolismul hemoglobinei.")
249
Dereglările catabolismului Hb
Icterul – ce se caracterizează prin: Hiperbilirubinemie - creşterea c% bilirubinei în sânge Coloraţia specifică a tegumentelor (galbena) şi lichidelor biologice
 şi lichidelor biologice.")
250
Cauzele hiperbilirubinemiei
Creşterea v de formare a bilirubinei (creşterea degradării hemului); Scăderea capacităţii ficatului de a capta bilirubina Scăderea capacităţii ficatului de a conjuga bilirubina Perturbarea mecanismului eliminării prin bilă Tulburări extrahepatice ale fluxului biliar
; Scăderea capacităţii ficatului de a capta bilirubina. Scăderea capacităţii ficatului de a conjuga bilirubina. Perturbarea mecanismului eliminării prin bilă. Tulburări extrahepatice ale fluxului biliar.")
251
3 tipuri de icter Prehepatic – hemolitic
Hepatic (hepatocelular) –parenhimatos Posthepatic – obstructiv
 –parenhimatos. Posthepatic – obstructiv.")
252
Tipuri de ictere A. hemolitic Exces de hemoliză
bilirubina neconjugată (în sînge) bilirubina conjugată (se elimină cu bila) B. Hepatic bilirubina neconjugată (în sînge) bilirubina conjugată C. Obstructiv bilirubina neconjugată (în sînge) bilirubina conjugată
 bilirubina conjugată. (se elimină cu bila) B. Hepatic. bilirubina neconjugată. (în sînge) bilirubina conjugată. C. Obstructiv. bilirubina neconjugată. (în sînge) bilirubina conjugată.")
253
Icterul hemolitic - prehepatic
Cauza – hemoliza masivă (degradarea exagerată a eritrocitelor) însoţit de o creştere a bilirubinei libere (indirecte), care depăşeşte capacitatea de conjugare a ficatului. Pe contul ei creşte şi bilirubina totală.
 însoţit de o creştere a bilirubinei libere (indirecte), care depăşeşte capacitatea de conjugare a ficatului. Pe contul ei creşte şi bilirubina totală.")
254
Icterul hepatic (hepatocelular)
Premicrosomal Microsomal postmicrosomal

255
Hepato premicrosomal Sindrom Defect Bilirubina Clinic
Crigler-Najjar I - AR Incapacitatea ficatului de a produce UDP-glucuroniltransferazei Bilirubinei indirecte Bilirub directă-abs în bilă Icter profund Dereglări SNC Crigler-Najjar II - AD Defect parţial UDP-glucuroniltransferazei Bilirubinei indirecte Semnele bolii sunt mai puţin severe

256
3. Boala Gilbert Cauzele: Legarea atipică mai stabilă a bilirubinei la albuminele sanguine; deficienţa captării bilirubinei libere de către ficat C% bilirubinei indirecte , fără modificări în urină şi masele fecale 4. Icterul neonatal “fiziologic”- hemoliza eritrocitelor +”imaturitatea” ficatului de a prelua, conjuga şi excreta bilirubina (deficit de UDP- glucoronat): C% bilirubinei indirecte
: C% bilirubinei indirecte.")
257
Hepatic microsomal- hepatocelular
Apare în hepatite acute virale, infecţioase; cronice, alcoolice, medicamentoase şi ciroze hepatice. Provocat de afectarea principalelor funcţii hepatice privind metabolismul pigmenţilor biliari (captarea, conjugarea şi excreţia) Creşte bilirubina totală, directă, indirectă; urobilinogenul în urină
 Creşte bilirubina totală, directă, indirectă; urobilinogenul în urină.")
258
Hepatic postmicrosomal
Provocat de perturbarea eliminării bilirubinei în bilă şi colestazei exclusiv intrahepatice A. Ereditar: 1. Sd Dubin-Johnson – defect al secreţiei bilirubinei conjugate în canaliculele biliare (biliubina conjugată mărită, culoarea ficatului este închisă) 2. Sd Rotor – asemănător cu Sd Dubin-Johnson, dar nu se produce sinteza pigmentului brun în ficat B. Dobândit – ciroza postnecrotică, biliară; atrezie canaliculă biliară
 2. Sd Rotor – asemănător cu Sd Dubin-Johnson, dar nu se produce sinteza pigmentului brun în ficat. B. Dobândit – ciroza postnecrotică, biliară; atrezie canaliculă biliară.")
259
Hepatic postmicrosomal
Creşte c% E, ce denotă colestaza (fosfotaza alcalină, gama glutamiltransferaza, 5-nucleotidaza)+ creşterea bilirubinei directe şi indirecte, urobilinogenul urinar micşorat sau lipseşte, dar se detectează pigmenţi şi săruri biliare
+ creşterea bilirubinei directe şi indirecte, urobilinogenul urinar micşorat sau lipseşte, dar se detectează pigmenţi şi săruri biliare.")
260
Icter posthepatic – mecanic - obstructiv
Tulburarea fluxului biliar – perturbarea eliminării bilei (blocarea canalelor biliare) Cauzele: Litiaza biliară Neoplasme (căi biliare) 1. Creşte f mult bilirubina directă (conjugată) – în sânge cât şi în urină - mai mare ca în icterul hepatic; 2. cresterea sarurilor biliare, 3. hipercolesterolemie, 4. Urobilina şi urobilinogenul vor fi normale sau scazute (în caz de obstrucţie totală – scaunul este decolorat).
 Cauzele: Litiaza biliară. Neoplasme (căi biliare) 1. Creşte f mult bilirubina directă (conjugată) – în sânge cât şi în urină - mai mare ca în icterul hepatic; 2. cresterea sarurilor biliare, 3. hipercolesterolemie, 4. Urobilina şi urobilinogenul vor fi normale sau scazute (în caz de obstrucţie totală – scaunul este decolorat).")
261
Icterele cu patogenie mixta
în afară de faptul că un icter mecanic poate duce la leziuni hepatocelulare, ca un icter prin hepatita poate determina trombi biliari sau ca un icter hemolitic poate avea si cauze inflamatorii hepatice si elemente obstructive, exista unele ictere care de la bun inceput au o patogenie mixta. Asa este cazul in toxiinfectiile grave (septicemia cu B. perfringens, spirochetoza icterohemoragica, pneumonia grava etc), in care icterul este hepatic si totodata hemolitic.
, in care icterul este hepatic si totodata hemolitic.")
Παρόμοιες παρουσιάσεις



















>")








