Κατέβασμα παρουσίασης
1
MALFORMAŢII CONGENITALE DE CORD LA COPII
Marcu Rudi, prof. universitar, Catedra Pediatrie Nr. 1 USMF “Nicolae Testemiţanu”

2
MCC MCC sunt anomalii structurale ale cordului cauzate de diferiţi factori nocivi exogeni şi endogeni în perioada embrionară de dezvoltare, în primele 2-8 săptămîni sau pînă la 3 luni de graviditate, atunci cînd are loc morfogeneza cordului.

3
PROBLEMELE ACTUALE ÎN CARDIOLOGIE PEDIATRICĂ
Alocarea aparatajului modern; Constatarea mică a MCC în perioada prenatală; Diagnosticarea joasă a MCC în maternitate; Diagnosticul precoce la prima etapă – medicina primară;

4
Efectuarea programului de Stat a MCC;
Tratamentul chirurgical cu întîrziere, mai ales la copii pînă la 1 an; Lipsa conlucrării medicilor în echipă; Cultura medicală joasă a societăţii; Cultura medicală a familiei; Probleme sociale.

5
11. În ţările dezvoltate BCC reprezintă o problemă prioritară pentru sănătatea publică;
12. Progresele rapide ale tehnicilor de diagnostic şi tratament au schimbat dramatic destinul bolnavilor şi familiilor acestora în ultimii 50 ani;

6
13. Durata şi calitatea vieţii a copiilor cu MCC trataţi chirurgical;
Au apărut probleme noi: 13. Durata şi calitatea vieţii a copiilor cu MCC trataţi chirurgical; 14. Încadrarea lor socială (oportunitatea sarcinilor, riscul recurenţei bolii); 15. Problema asigurărilor medicale.
; 15. Problema asigurărilor medicale.")
7
MCC. Incidenţa După OMS este de la 1000 de copii nou-născuţi. În mediu 10 copii la 1000 nou născuţi. MCC ocupă 25-50% din toate malformaţiile congenitale. În Republica Moldova cifra se estimează aproape la acelaşi nivel şi anual în prezent se nasc în jur de 500 de copiii cu MCC, neluînd în seamă că natalitatea s-a micşorat vădit.

8
MCC. Etiologia Factorii genetici (cromosomiali şi genetici) 10-12%.
Influenţa factorilor de mediu 2% Teoria multifactorială – interacţiunea între factorii genetici intrinseci şi factorii de mediu 85-90% Factorii genetici: în majoritatea cazurilor se remarcă o incidenţă crescută a MCC cu sindroame genetice: în jur de 9% din copiii cu MCC sunt purtătorii unui sindrom DOWN. Asocieri recunoscute sunt şi în sindroamele NOONAN, TURNER, WILLIAMS, MARFAN, HOLT ORAM şi a.

9
Anomalii extracardiace Anomalii cardiace
Sindroame Anomalii extracardiace Anomalii cardiace Holt-Oram Anomalia genetică este situată pe cromosomul 12, Se transmite autosomal-dominant Anomalii a membrelor superioare: hipolazia eminenţei tenarului; Comunicaţia interatrială de tip osteum secundum; Bloc atrio-ventricular de gr. I Ellis-van Creveld Nanism, hexadactilie Canal atrioventricular Auricul unic Rubinstein-Taybi Facies particular Retard statural Retard mental Canal arterial Anomalia arcului aortei Mai rar CIA, CIV

10
Epifiză punctiformă Nanism, accentuarea spinei nazale Comunicarea interventriculară, Anomalia Fallot Cornelia de Lange Dismorfie facială Ţunt stînga-dreaptă MULIBREY Hipertelorism Pericardită constrictivă Noonan Retard staturo-ponderal Dismorfie facială şi toracică Stenoza pulmonară Hipertrofia septală (miocardiopatie hipertrofică) Comunicaţia interatrială (osteum secundum)
 Comunicaţia interatrială (osteum secundum)")
11
Goldenhar Malformaţii de urechi şi ochi Hemivertebre Malformaţii renale Tetralogia Fallot Di George Dismorfie facială Hipoplazia timusului şi paratiroidei (deleţia cromosomului 22) Trunchi arterial comun Atrezia pulmonară asociată cu CIV, Fallot, anomalii ale arcului aortic Pierre Robin Retrognatism Dehiscenţa palatului Comunicaţie interventriculară Velocardiofacial Voce nazonată (crom. 22) Comunicaţii interventriculare Fallot
 Trunchi arterial comun. Atrezia pulmonară asociată cu CIV, Fallot, anomalii ale arcului aortic. Pierre Robin. Retrognatism. Dehiscenţa palatului. Comunicaţie interventriculară. Velocardiofacial. Voce nazonată (crom. 22) Comunicaţii interventriculare. Fallot.")
12
Cayler Asimetrie facială Comunicaţie interventriculară Fallot Alagille Dismorfie facială Colestaza cronică, asociată cu hipoplazia canalelor biliare intrahepatice Facies caracteristic Anamolii vertebrale Hipoplazia sau stenoza ramurilor pulmonare CIV CHARGE Atrezia coanelor Colobome Surditate Anomalii genitale Retard mental Şunt stînga dreapta

13
Williams-Beuren Dismorfie facială Retard mental (crom. 7) Stenoza supravalvulară aortică Şi stenoză de trunchi şi de ramuri pulmonare Fanconi Anomalia degetelor şi antebraţului, anemie Canal arterial Coarctaţie Fallot

14
Asocierea MCC cu sindroame cromozomiale
se întîlneşte la 15% dintre noi-născuţi, dar nici un tip de malformaţie nu este patognomică pentru un anumit sindrom cromozomial Este bine cunoscută incidenţa crescută a canalului atrio-ventricular în sindromul DOWN - pînă la 40% şi a leziunilor obstructive ale cordului stîng în sindromul TURNER.

15
Transmiterea autozomal dominantă este demonstrată în sindroamele MARFAN, WILLIAMS şi HOLT ORAM, iar transmiterea autozomal recisivă în sindromul ELLIS van CREVELD. Mutaţia genetică reprezintă cauza unor malformaţii cardiace ce apar atît izolate, cît şi însoţite de malformaţii extracardiace (sindroame).
.")
16
Sunt implicaţi izolat în apariţia
Factorii de mediu Sunt implicaţi izolat în apariţia MCC doar în 2-5%. Ei pot fi: Viruşii (rubeola); Diabetul matern - constituie un risc major pentru tetrada Fallot sau trunchi arterial comun; Folosirea unor medicamente – cu litiu, amfetamine, anticonvulsivante sau tranchilizante; Fumatul şi consumul de alcool – creşte incidenţa defectului septal, alte substanţe toxice; radiaţiile, hipoxia etc.
; Diabetul matern - constituie un risc major pentru tetrada Fallot sau trunchi arterial comun; Folosirea unor medicamente – cu litiu, amfetamine, anticonvulsivante sau tranchilizante; Fumatul şi consumul de alcool – creşte incidenţa defectului septal, alte substanţe toxice; radiaţiile, hipoxia etc.")
17
Teoria multifactorială în 85% cazuri şi are loc atunci cînd:
Este predispoziţie genetică Perioada vulnerabilă de dezvoltare embrionară 2-8 săptămâni – 3 luni Influenţa factorilor de mediu în această perioadă de dezvoltare embrionară şi predispoziţia genetică.

18
Cardiopatii secundare unor patologii materne
Etiologie Cardiopatii Infecţioasă Rubeola Canal, stenoză pulmonară Etiologie toxică Antiepileptice Litiu Alcool Antiprostoglandine Diverse Anomalia Ebstein CIV, CIA Miocardiopatia Ventricolului drept cu închiderea canalului în uter. Cauze materne Diabet Fenilcetonurie CIV, transpoziţie, hipo-ventricol stîng Fallot, CIV, Canal

19
Diagnosticul prenatal al MCC
Constituie o importanţă deosebită în prevenţia naşterii nou-născuţilor, mai ales cu MCC grave. Perioada ideală de a depista MCC la făt este de 18 – săptămîni de sarcină. Cu ajutorul ecocardiografiei fetale pot fi depistate pînă la 80 – 90% din MCC la făt.

20
Clasificarea patofiziologică a MCC conform dereglărilor hemodinamice în circuitul pulmonar
Dereglările hemodinamice Fără cianoză Cu cianoză Cu îmbogăţirea circuitului pulmonar PCA, DSA, DSV, CAV Complexul Eisenmenger, TGA, TAC Cu reducerea circuitului pulmonar Stenoza a. pulmonare Tetrada Fallot, atrezia v.tricuspide, TGA cu stenoza AP, TAG fals, B. Ebstein Cu schimbări în circuitul sistemic Stenoza Ao, coarctaţia de Ao Fără dereglări hemodinamice Dextrocardia, anomalia arc. Ao, DSV mic

21
Clasificarea patogenică a MCC (Moss şi Adams, a. 1996)
Comunicarea anormală între circulaţia sistemică şi pulmonară (MCC cu şunt stînga-dreapta): DSA, DSV, CAV, PCA Anomalii ale tractului de ieşire din VS: stenoza Ao valvulară, stenoza Ao supravalvulară, Sd. Williams, coarctaţia de Ao, Sd. de cord stîng hipoplastic Anomalii ale tractului de ieşire din VD: stenoza pulmonară valvulară izolată, stenoza ramurilor AP, atrezia AP, tetrada Fallot.
: DSA, DSV, CAV, PCA. Anomalii ale tractului de ieşire din VS: stenoza Ao valvulară, stenoza Ao supravalvulară, Sd. Williams, coarctaţia de Ao, Sd. de cord stîng hipoplastic. Anomalii ale tractului de ieşire din VD: stenoza pulmonară valvulară izolată, stenoza ramurilor AP, atrezia AP, tetrada Fallot.")
22
Anomalii ale valvelor atrioventriculare: MCC ale valvei mitrale - stenoza mitrală congenitală, MC ale valvei tricuspide - atrezia valvei tricuspide, anomalia Ebstein. Originea anormală a marilor vase şi arterelor coronare: transpoziţia completă şi corectată a vaselor mari, trunchiul arterial comun, originea anormală a arterelor coronare. Anomalia de întoarcere a circulaţiei venoase pulmonare: anomalia parţială şi totală de întoarcere venoasă pulmonară. Malpoziţia cordului şi situsului visceral.

23
Clasificarea după Park M.K., 2009
PALIDE: A. Leziuni valvulare şi vasculare obstructive fără şunt asociat: obstrucţia tractului de ejecţiei VS: stenoză subaortică; stenoză aortică valvulară; stenoză aortică supravalvulară. Co Ao; întreruperea arcului aortic; stenoză pulmonară cu sept ventricular intact. B. ŞUNT STÂNGA-DREAPTA: DSV; DSA; CAV; CAP.

24
Cianotice A. şunt dreapta stânga: atrezia a.pulmonare;
cale dublă de ieşire din VD; anomalia Ebştein; tetralogia Fallot. B. vicii complexe: transpoziţia vaselor magistrale; drenaj venos aberant total; MCC cu hemodinamica univentriculară.

25
Diagnosticul general al MCC
Diagnosticul MCC trebuie să fie un diagnostic precoce! Anamneza: antecedente obstetricale, afecţiuni virotice şi toxice, în special în săptămînile 6-8 de sarcină, leziuni malformative la fraţi, sindroame dis- morfice şi ereditare în cadrul antecedentelor colaterale), Semne funcţionale: dispnee de efort, infecţii bronhopulmonare repetate, transpiraţii, întîrziere în dezvoltarea pondero-staturală, durere precordială de tip anginos, cefalee).
, Semne funcţionale: dispnee de efort, infecţii bronhopulmonare repetate, transpiraţii, întîrziere în dezvoltarea pondero-staturală, durere precordială de tip anginos, cefalee).")
26
Examenul obiectiv. Inspecţia generală:
Cianoza precoce sau tardivă Crize de cianoză paroxistică, Hipocratism digital

27
Diagnosticul paraclinic:
Radiologia toracică, Electrocardiografia, Ecocardiografia Doppler Investigaţii speciale: Cateterism cardiac, Angiocardiografie, Rezonanţă magnetică nucleară, Tomografia computerizată.

28
Este determinat de trei factori de bază:
Tabloul clinic Este determinat de trei factori de bază: Particularităţile anatomice ale malformaţiei Gradul de compensare Complicaţii

29
Fazele evolutive MCC 1. Etapa de adaptare, caracterizată prin capacitatea organismului copilului de a se acomoda dereglărilor hemodinamice existente (variază ca durată şi manifestări clinice); 2. Etapa de compensare relativă (acuze, semne clinice practic lipsesc, se ameliorează dezvoltarea fizică); 3. Etapa terminală, ce apare la epuizarea posibilităţilor compensatorii şi dezvoltarea proceselor distrofice, ireversibile în muşchiul cardiac şi organele parenhimatoase cu prognostic întunecos şi diverse complicaţii.
; 2. Etapa de compensare relativă (acuze, semne clinice practic lipsesc, se ameliorează dezvoltarea fizică); 3. Etapa terminală, ce apare la epuizarea posibilităţilor compensatorii şi dezvoltarea proceselor distrofice, ireversibile în muşchiul cardiac şi organele parenhimatoase cu prognostic întunecos şi diverse complicaţii.")
30
Incidenţa DSA – 6-10% DSV – 20% PCA – 5 -10% CAV – 4-5%
Fereastra aorto-pulmonară 2% Complex Eisenmenger 1-2% Transpoziţia de vase mari şi trunchi arterial comun dirijat – 1%

31
MCC cu şunt stînga-dreapta
Au dereglări hemodinamice comune, cînd în circuitul mic pătrunde un volum de sînge mai mare. Particularităţile clinice sunt determinate de dezvoltarea hipervolemiei şi hipertensiei în circuitul mic şi insuficienţa cardiacă precoce, predispoziţie la frecvente şi repetate pneumonii sau infecţii respiratorii.

32
Grupul de MCC cu şunt stânga-dreapta
Intracardiace: ♯ DSV ♯ DSA ♯ CAV Extracardiace: ♯ CAP ♯ fistula aorto-pulmonară ♯AP dreapta originară din Ao

33
Defect septal ventricular
20-30% din toate CC, locul 2 după bicuspidia A Incidenţa: 1,5-3,5 ; 4,5-7/1000 În 25-40% cazuri – închidere spontană până la vârsta de 2 ani axioma “ suflu mare = defect mic” nu este aplicabila în toate cazurile!!!!!!! 1879 Roger ( DSV mic) examenul clinic cu depistarea unui suflu cardiac caracteristic de DSV asociat aberaţii cromosomiale (trisomia 13,trisomia18, trisomia21) Dar 95% din pacienţi cu DSV nu au anomalii genetice
 examenul clinic cu depistarea unui suflu cardiac caracteristic de DSV. asociat aberaţii cromosomiale. (trisomia 13,trisomia18, trisomia21) Dar 95% din pacienţi cu DSV nu au anomalii genetice.")
34
4 parţi componente al SIV
20% - muscular 5% - subaortal 5% - reg. de joncţiune dintre VM şi VT 70-80% - membranos;

35
EcoCG Transesofagiană
Clasificare DSV (2) Estimarea ecocardiografică DSV mic DSV de dimensiuni medii DSV mare (larg) Risc de sub- /supraestimare EcoCG Transesofagiană
 Estimarea ecocardiografică. DSV mic. DSV de dimensiuni medii. DSV mare (larg) Risc de sub- /supraestimare. EcoCG Transesofagiană.")
36
Fiziopatologia în DSV Şunt stânga-dreapta: 1. Supraîncarcare
de volum VS (dilatare / hipertrofie VS) 2. Hipervolemie în circuitul vascular pulmonar (HTAP) 3. Reducerea debitului cardiac sistemic (ICC)
 2. Hipervolemie în circuitul. vascular pulmonar (HTAP) 3. Reducerea debitului. cardiac sistemic (ICC)")
37
Evoluţia naturală I - Închidere spontană (2-4 ani): muscular (80%)
perimembranos (35-40%) DSV infundibular practic nu se închide spontan II – Complicaţii: ICC, Sindrom Eisenmenger, IAo, III – Moarte subită
 DSV infundibular practic nu se închide spontan. II – Complicaţii: ICC, Sindrom Eisenmenger, IAo, III – Moarte subită.")
38
Tratamente chirurgicale
Inchiderea chirurgicală cu patch

39
Dispozitive pentru inchiderea defectelor septale
40
Complicaţii postoperatorii: după corecţie chirurgicală în DSV
Defect rezidual, recurent HTAP sau IR (tulburari de ventilare pre- sau imediat postoperator BAV, BRD, aritmii ventriculare Disfuncţia VS Supraveghere anuală la cardiolog

41
Defect septal atrial predomină sexul feminin 2:1
frecvenţa circa 8% predomină sexul feminin 2:1 diagnostic antenatal ? adesea bine tolerat închiderea spontană este posibilă în primii 2 ani de viaţă pot fi în sindroame genetice ( Holt-Oram, Down)
")
42
DSA clasificare, tipuri
ostium secundum inalt; 15% În sp ostium primum jos; anom VM sinus venosus sinus coronarian

43
Suprâncărcarea (dilatare) HTAP cu evoluţie în fixă
Fiziopatologia DSA Este în funcţie de: importanţa şuntului marimea defectului vascularizarea pulmonară Suprâncărcarea (dilatare) diastolică ale VD şi AD accentuează care cu timpul dce la HTAP cu evoluţie în fixă la ani
 diastolică ale VD şi AD. accentuează. care cu timpul dce la. HTAP cu evoluţie în fixă. la ani.")
44
DSA mic Semnele clinice
un suflu discret cu dedublarea zg2 la focarul AP Supraveghere obişnuită în aşteptarea unei ocluzii intervenţionale prin cateterism

45
DSA de dimensiuni medii
Diagnosticul clinic: Asimptomatici /IR recurente Suflul sitolic 2/6 şi dedublarea zg2 la focarul AP Rx: ICT majorat puţin, hipervascularitare pulmonară ECG: Bloc de ram drept incomplet ECOCARDIOGRAFIA 2D, doppler

46
DSA de dimensiuni medii
Supravegherea anuală până la închidere Cu tratament medicamentos de susţinere la necesitate Chirurgical: La fetiţe varsta circa 15 ani (toracotomie submamară) La băeţi la 5 ani (sternotomie)
 La băeţi la 5 ani (sternotomie)")
47
DSA de dimensiuni mari Diagnostic clinic Malnutriţie moderată
Intoleranţă la efort IR recurente Suflul sistolic 2/6 Dedublarea zg2 la AP La focarul VT zg 1 accentuat, Rx: cardiomegalie, hipervolemie pulmonară ECG: Bloc de ram drept incomplet Ecocardiografia

48
Complicaţii postoperatorii în DSA
De regulă rezultat bun Sindrom postpericardiotomie Regurgitaţii valvulare Defect rezidual Embolii paradoxale Aritmii atriale

49
Se depistează prin semne clinice Rx toracelui, ecocardiografia, ECG
De reţinut!!!!!!! Şuntul stânga-dreapta reprezintă trecerea unei cantităţi de sânge saturat în cordul drept cu supraîncarcarea circuitului vascular pulmonar Se depistează prin semne clinice Pulmonare(tahipnee!HTAP fixată) Cardiace (IC iniţial pulmonară apoi globală)!!! Rx toracelui, ecocardiografia, ECG
 Cardiace (IC iniţial pulmonară apoi globală)!!! Rx toracelui, ecocardiografia, ECG.")
50
Tratamentul conservativ pre- şi postoperator:
1. Tratamentul hipertensiunii vasculare pulmonare cu IECA, diuretice (Furosemid, Hidroclortiazid) 2. IC cu tonicardice IECA (Captopril, Enalapril, Lizinopril); Digoxina, (Dopamina în cazuri de urgenţă), 3. Tratamentul complicaţiilor (tratament activ cu antibiotice a infecţiilor bacteriene). Tratamentul chirurgical: plastia defectului septal, reconstruirea valvelor în CAV, în PCA – secţiunea, nu ligatura canalului arterial, etc.
 2. IC cu tonicardice IECA (Captopril, Enalapril, Lizinopril); Digoxina, (Dopamina în cazuri de urgenţă), 3. Tratamentul complicaţiilor (tratament activ cu antibiotice a infecţiilor bacteriene). Tratamentul chirurgical: plastia defectului septal, reconstruirea valvelor în CAV, în PCA – secţiunea, nu ligatura canalului arterial, etc.")
51
Transpoziţia completă a vaselor mari dirijată
MCC caracterizată prin originea vaselor mari din ventricule neadecvate: Ao se naşte din VD şi AP din VS. Reprezintă 5-7% din MCC. Predominanţa sexului masculin (70%) În lipsa unui şunt, care să permită amestecul de sînge, viaţa nu este posibilă. Se asociază anomalia a. coronare.
 În lipsa unui şunt, care să permită amestecul de sînge, viaţa nu este posibilă. Se asociază anomalia a. coronare.")
52
Transpoziţia completă a vaselor mari simptomatologia
Este evidentă clinic din I zi de viaţă; Este cea mai frecventă cauză de cianoză cardiacă la nou-născut; Suflu sistolic, insuficienţă respiratorie majoră (tahipnee), IC, hipoxie cronică – factor de risc pentru enterocolita ulceronecrotică, accidente vasculare cerebrale precoce (hemipareză), retard mental, explicat de hipoxemia cronică severă.
, IC, hipoxie cronică – factor de risc pentru enterocolita ulceronecrotică, accidente vasculare cerebrale precoce (hemipareză), retard mental, explicat de hipoxemia cronică severă.")
53
Transpoziţia completă a vaselor mari tratamentul
Perfuzie cu prostaglandina E (menţinerea CAP deschis), tratarea acidozei metabolice; Septostomie cu balon pentru asigurarea unui şunt interatrial; Tratament chirurgical – transferul coronarelor (corecţie “anatomică”, tehnica Jatene).
, tratarea acidozei metabolice; Septostomie cu balon pentru asigurarea unui şunt interatrial; Tratament chirurgical – transferul coronarelor (corecţie anatomică , tehnica Jatene).")
54
MCC cu reducerea circuitului pulmonar

55
Tetralogia Fallot Incidenţa tetralogiei Fallot (TF) – 10% din MCC şi 70% din cele cianogene. Elementele TF sunt: Stenoza pulmonară: valvulară (25%), infundibulară (50%), asociate (25%), supravalvulară DSV larg situat sub valva dreaptă aortică. Aorta călare pe istm. Hipertrofia VD. Asocieri frecvente (40%) sunt: DSA, PCS, canal A-V, absenţa arterei pulmonare.
, infundibulară (50%), asociate (25%), supravalvulară. DSV larg situat sub valva dreaptă aortică. Aorta călare pe istm. Hipertrofia VD. Asocieri frecvente (40%) sunt: DSA, PCS, canal A-V, absenţa arterei pulmonare.")
56
Factori de compensare: poliglobulia, circulaţia, PCA.
Tetralogia Fallot Fiziopatologie fluxul dreapta-stânga în relaţie cu gradul stenozei rezistenţa vasculară sistemică mărimea DSV şi poziţia aortei Factori de compensare: poliglobulia, circulaţia, PCA.

57
Forme fiziopatologice şi clinice
Tetralogia Fallot Forme fiziopatologice şi clinice Forma cianotică: obstrucţie severă la golirea VD cu flux pulmonar redus şi creşterea fluxului de la VD la Ao şi VS cu hipoxie, cianoză severă şi policitemie. Forma TF acianotică: obstrucţie redusă cu şunt dreapta-stânga mic şi şunt mai crescut stânga-dreapta (VSVD). Starea clinică a acestei forme este uşoară. Forma TF de “pseudotrunchi” – se întâlneşte în atrezia pulmonară cu şunt mare dreapta-stânga cu cianoză şi hipoxie marcată. Perfuzia pulmonară este redusă şi este asigurată în mod limitat de venele bronşice sau PCA. Starea clinică severă şi mortalitatea este crescută.
. Starea clinică a acestei forme este uşoară. Forma TF de pseudotrunchi – se întâlneşte în atrezia pulmonară cu şunt mare dreapta-stânga cu cianoză şi hipoxie marcată. Perfuzia pulmonară este redusă şi este asigurată în mod limitat de venele bronşice sau PCA. Starea clinică severă şi mortalitatea este crescută.")
58
Tetralogia Fallot Tabloul clinic
Majoritatea sunt simptomatici cu cianoză şi hipoxie ce apare înainte de vârsta de 1 an după naştere. Sunt remarcate: dispnee, angină pectorală, cefalee, ameţeli, poziţie pe vine, crize cu hipercianoză (hiperpnee, convulsii, sincopă, accidente cerebrovasculare şi decese) şi, mai rar, fenomene de insuficienţă cardiacă şi palpitaţii prin tulburările de ritm, în special tahicardii ventriculare.
 şi, mai rar, fenomene de insuficienţă cardiacă şi palpitaţii prin tulburările de ritm, în special tahicardii ventriculare.")
59
Tetralogia Fallot Complicaţii: Datele fizice: aritmii, cianoză,
tromboze, ictus, embolii paradoxale, abces cerebral, endocardită infecţioasă, insuficienţă VD, moarte subită. Datele fizice: cianoză, hipocratism, freamăt sistolic parasternal stâng cu pulsaţia VD. La auscultaţie – zgomot II unic şi redus la pulmonară, suflu parasternal în spaţiul III-IV prin DSV.

60
Tetralogia Fallot Examinări
Examene de laborator: hematocritul şi hemoglobina (crescute), coagulograma. ECG: AE deviată spre dreapta, hipetrofia VD, aritmii (fibrilaţie atrială, tahicardii ventriculare). Examen Rx cardiopulmonar: cord normal sau mărit uşor, “inima în sabot”, vascularizaţie pulmonară redusă. Echo-CG 2D: DSV, SP, hipertrofie VD, poziţia călare a aortei. Cateterismul şi cardioangiografia: determină presiunea crescută în VD, gradientul şi localizarea SP, gradul hipoxiei în VS şi aortă, circulaţia bronşică, poziţia aortei.
, coagulograma. ECG: AE deviată spre dreapta, hipetrofia VD, aritmii (fibrilaţie atrială, tahicardii ventriculare). Examen Rx cardiopulmonar: cord normal sau mărit uşor, inima în sabot , vascularizaţie pulmonară redusă. Echo-CG 2D: DSV, SP, hipertrofie VD, poziţia călare a aortei. Cateterismul şi cardioangiografia: determină presiunea crescută în VD, gradientul şi localizarea SP, gradul hipoxiei în VS şi aortă, circulaţia bronşică, poziţia aortei.")
61
Tetralogia Fallot Evoluţie
Mortalitatea este crescută, 30% în primul an, 50% la 3 ani, 75% la 10 ani. Această evoluţie este modificată favorabil prin intervenţii chirurgicale timpurii. Bolnavii cu forma acianotică pot atinge vârsta maturităţii fără complicaţii.

62
Tetralogia Fallot Tratament
Acces de rău hipoxic: poziţie pe vine, se administrează O2, propranolol i/v (0,01-0,25 mg/kg) încet, bicarbonat de sodiu 1 mEq/kg i/v repetat la 10-15´, şi, rar, morfină 0,2 mg/kg subcutan sau i/m. Accesele cu hipercianoză: pot fi prevenite cu β adrenoblocante per os 0,5-1,5 mg/kg fiecare 6 ore. Profilaxia endocarditei bacteriene. Tratamentul anemiei.
 încet, bicarbonat de sodiu 1 mEq/kg i/v repetat la 10-15´, şi, rar, morfină 0,2 mg/kg subcutan sau i/m. Accesele cu hipercianoză: pot fi prevenite cu β adrenoblocante per os 0,5-1,5 mg/kg fiecare 6 ore. Profilaxia endocarditei bacteriene. Tratamentul anemiei.")
63
Indicaţiile de intervenţie chirurgicală
Tetralogia Fallot Indicaţiile de intervenţie chirurgicală copii cu cianoză, hipoxie crize cu hipercianoză hematocrit peste 65% Operaţii paliative (Blalock-Taussing): şunt aortă a. pulmonară, ce creşte fluxul pulmonar. Persistă pericol de complicaţii (embolii, endocardită infecţioasă, boală vasculară pulmonară obstructivă, şunt dreapta-stânga). La 3-5 ani: corecţie totală – valvotomie pulmonară, închiderea DSV şi conduct VD-AP în caz de atrezie pulmonară.
: şunt aortă a. pulmonară, ce creşte fluxul pulmonar. Persistă pericol de complicaţii (embolii, endocardită infecţioasă, boală vasculară pulmonară obstructivă, şunt dreapta-stânga). La 3-5 ani: corecţie totală – valvotomie pulmonară, închiderea DSV şi conduct VD-AP în caz de atrezie pulmonară.")
64
Incidenţa - 1% din totalul MCC.
Boala Ebştein Incidenţa - 1% din totalul MCC. Morfopatologie: ataşarea anormală a valvei anterioare şi posterioare a tricuspidei la peretele VD, valva tricuspidă deplasată şi displazică cu stenoză sau insuficienţă, dilatarea AD şi reducerea dimensiunii VD. Asocierea altor anomalii este frecventă: DSA (50%), SP, DSV, TMV.
, SP, DSV, TMV.")
65
Boala Ebştein Fiziopatologie
insuficienţa valvei tricuspide cu creşterea presiunii în VD; şunt dreapta-stânga prin DSA sau deschiderea foramen ovale (75%); reducerea funcţiei VD şi a fluxului pulmonar; cianoză cu hipoxie variabilă; aritmii.
; reducerea funcţiei VD şi a fluxului pulmonar; cianoză cu hipoxie variabilă; aritmii.")
66
Boala Ebştein evoluează asimptomatic la mulţi pacienţi.
Tabloul clinic Boala Ebştein evoluează asimptomatic la mulţi pacienţi. Simptomele iniţiale: dispnee, oboseală la efort, palpitaţii, sincope rare prin aritmii, cianoză prin şunt dreapta-stânga (DSA). Datele fizice: suflu sistolic parasternal şi la tricuspidă, suflul diastolic în aceeaşi localizare. Aritmiile: tahicacardia paroxistică supraventriculară (25%), ventriculară (20%). Complicaţii: sincope, insufcienţă cardiacă congestivă, embolii, abces cerebral, moarte subită (20%).
. Datele fizice: suflu sistolic parasternal şi la tricuspidă, suflul diastolic în aceeaşi localizare. Aritmiile: tahicacardia paroxistică supraventriculară (25%), ventriculară (20%). Complicaţii: sincope, insufcienţă cardiacă congestivă, embolii, abces cerebral, moarte subită (20%).")
67
Boala Ebştein Investigaţii
Radiologic: cordul este mărit, forma “mingei de rugby” prin AD lărgit, vascularizarea pulmonară este redusă. Echo-CG: VD mic, mişcare paradoxală a septului interventricular, excursie mărită a valvei tricuspide anterioare. Evoluţia: o mare parte de pacienţi au o evoluţie bună, până la ani. Decesul are loc prin insuficienţă cardiacă, debit cardiac scăzut, aritmii severe.

68
Boala Ebştein Tratament
Tratament medical, controlul aritmiilor şi al insuficienţei cardiace. Tratament chirurgical este indicat la pacienţi cu simptome relativ severe, cardiomegalie, cianoză şi hipoxie. Metodele chirurgicale utilizate sunt: anuloplastia valvei tricuspide, procedeul Gleen (anastomoza venei cave superioare cu artera pulomanră dreaptă), proteză la VT, închiderea DSA.
, proteză la VT, închiderea DSA.")
69
Stenoza pulmonară valvulară izolată
Reprezintă 8-10% din MCC SP cu valva displastică este întîlnită la copii cu s-ul Noonan (40%). Simptomatologie: pacientul poate fi asimptomatic şi doar la o SP critică apar semne clinice. În debit cardiac insuficient – dispnee de efort, oboseală. În SP strînse – sincope la efort. Suflu sistolic şi accentul zg. II la AP
. Simptomatologie: pacientul poate fi asimptomatic şi doar la o SP critică apar semne clinice. În debit cardiac insuficient – dispnee de efort, oboseală. În SP strînse – sincope la efort. Suflu sistolic şi accentul zg. II la AP.")
70
Stenoza pulmonară valvulară izolată
Tratamentul: Se practică valvuloplastia cu balon Valvulotomia chirurgicală pe cord deschis– în SP critică (stenoza combinată). Valvuloplastia transluminală percutanată cu balon – metodă terapeutică de elecţie. Tratamentul conservativ: în caz de IC este contraindicat tratamentul digitalo-diuretic, xantinele, se administrează β adrenoblocantele (metoprolol, propranolol, atenolol),
. Valvuloplastia transluminală percutanată cu balon – metodă terapeutică de elecţie. Tratamentul conservativ: în caz de IC este contraindicat tratamentul digitalo-diuretic, xantinele, se administrează β adrenoblocantele (metoprolol, propranolol, atenolol),")
71
Stenoza pulmonară valvulară izolată
Complicaţii: Insuficenţă cardiacă Endocardită bacteriană atât înainte, cât şi după intervenţie chirurgicală. Moarte subită

73
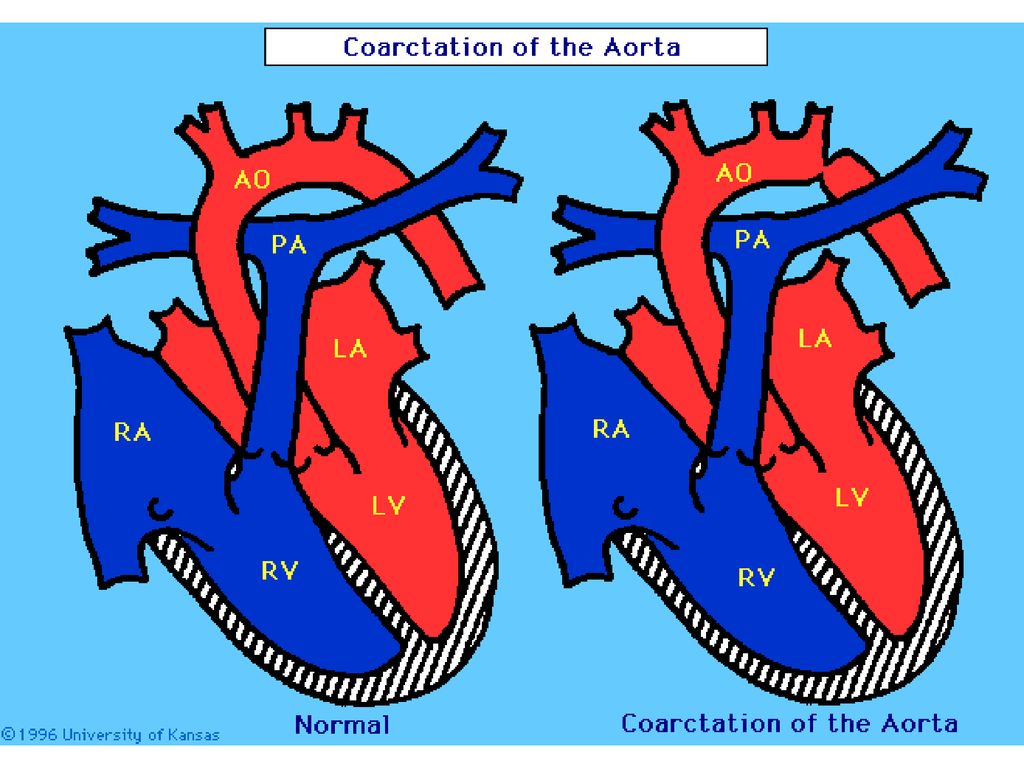
Coarctaţia de aortă simptomatologia
Deosebim 3 aspecte clinice majore: Sugar cu IC (debut precoce, insuficienţă cardiacă catastrofală, diagnostic dificil, evoluţie severă) Copil cu HTA (discrepanţă între valorile TA de la membrele superioare şi inferioare, puls femural absent sau slab, suflu sistolic de ejecţie, evoluţie mai moderată, prognostic favorabil) Copil cu suflu sistolic (fără semne clinice majore, în CoA largă)
 Copil cu HTA (discrepanţă între valorile TA de la membrele superioare şi inferioare, puls femural absent sau slab, suflu sistolic de ejecţie, evoluţie mai moderată, prognostic favorabil) Copil cu suflu sistolic (fără semne clinice majore, în CoA largă)")
74
Tipurile de coartaţie Co Ao tip adult;
Co Ao tip infantil (hipoplazia Ao);
;")
75
Coarctaţia de aortă Complicaţiile: IC, anevrism sau ruptura de aortă,
endocardită bacteriană, hemoragie cerebrală, leziuni aterosclerotice precoce. Tratamentul: Rezecţia zona ingustată Anastomoza termino-terminală Conservativ se prevede tratamentul IC şi a HTA

76
Profilaxia şi supravegherea copiilor cu MCC
Profilaxia endocarditei bacteriene pre- şi postoperator (antibiotice se recomandă înaintea oricărei manevre invazive care poate determina bacteriemie – intervenţii ORL, stomatologice, gastroscopice, cistoscopice, incizia unui abces, în implantarea de valvule artificiale sau peace-maker); Vaccinările, intervenţiile stomatologice şi ORL nu sunt contraindicate; Infecţiile intercurente vor fi tratate enrgic cu antibiotice;
; Vaccinările, intervenţiile stomatologice şi ORL nu sunt contraindicate; Infecţiile intercurente vor fi tratate enrgic cu antibiotice;")
77
Activitatea fizică va fi limitată numai în MCC severe, cu şunt important;
Activitatea şcolară, ambianţa psihică şi măsurile de ordin pedagogic trebuie adaptate conform substratului morfoclinic al afecţiunii; Pacienţii trebuie să fie supravegheaţi de către medicul de familie şi regulat – odată la luni, în dependenţă de tipul MCC să fie trimişi la specialistul cardiolog pediatru.

78
II. Anomalii ale tractului de ieşire din ventriculul stâng
a. Stenoza aortică valvulară Definiţia Stenoza aortică (SA) valvulară = tulburare de ejecţie a VS ca urmare a strâmtorării orificiului valvular aortic. Leziuni anatomice: Valve aortice îngroşate şi unite prin marginile libere realizând un "dome" cu orificiul central stenotic, inelul valvular este subdezvoltat, hipertrofia VS, ± dilataţia poststenotică a aortei ascendente.
 valvulară = tulburare de ejecţie a VS ca urmare a strâmtorării orificiului valvular aortic. Leziuni anatomice: Valve aortice îngroşate şi unite prin marginile libere realizând un dome cu orificiul central stenotic, inelul valvular este subdezvoltat, hipertrofia VS, ± dilataţia poststenotică a aortei ascendente.")
79
Iniţial se produce hipertrofia VS,
Hemodinamica Factorul determinant al tulburărilor hemodinamice este gradientul de presiune dintre VS şi Ao. Iniţial se produce hipertrofia VS, în timp suprasolicitarea duce la instalarea insuficienţei VS cu stază retrogradă. Consecutiv instalării insuficienţei se produce şi o reducere a fluxului sanguin în arterele coronare.

80
Pulsul periferic slab bătut orientează spre stenoze aortice severe.
Tabloul clinic Ascultatoric : suflu sistolic intens, aspru, localizat în spaţiul II intercostal drept iradiază pe traiectul arterelor carotide şi se opreşte înainte de zgomotul II (este deci protomezositolic). Se asociază cu freamăt ce se palpează în furculiţa sternală. Pulsul periferic slab bătut orientează spre stenoze aortice severe. În timp : scade toleranţa la efort, apar sincope sau lipotimii de efort
. Se asociază cu freamăt ce se palpează în furculiţa sternală. Pulsul periferic slab bătut orientează spre stenoze aortice severe. În timp : scade toleranţa la efort, apar sincope sau. lipotimii de efort.")
81
aorta dilatată (poststenotic) sau
Radiologic : cord cu dimensiuni normale ± vârful rotunjit (semn de hipertrofie ventriulară stângă), aorta dilatată (poststenotic) sau stază retrogradă pulmonară (supraîncărcare vasculară pulmonară) EKG normal în formele comune şi uneori chiar în SA strânse. În formele severe se decelează: HVS, subdenivelarea ST şi inversarea undei T în derivaţiile precordiale. Ecocardiografia: evidenţiază stenoza aortică subvalvulară, hipertrofia ventricului stâng şi dimensiunea VS la sfârşitul diastolei ! Tratamentul este chirurgical. Alegerea procedeului : valvuloplastie cu balon, valvulotomia, înlocuirea valvei aortice cu o valvă prostetică ţine în primul rând de gradientul transstenotic.
, aorta dilatată (poststenotic) sau. stază retrogradă pulmonară (supraîncărcare vasculară pulmonară) EKG. normal în formele comune şi uneori chiar în SA strânse. În formele severe se decelează: HVS, subdenivelarea ST şi. inversarea undei T în derivaţiile precordiale. Ecocardiografia: evidenţiază. stenoza aortică subvalvulară, hipertrofia ventricului stâng şi. dimensiunea VS la sfârşitul diastolei ! Tratamentul este chirurgical. Alegerea procedeului : valvuloplastie cu balon, valvulotomia, înlocuirea valvei aortice cu o valvă prostetică ţine în primul rând de gradientul transstenotic.")
82
b. Stenoza aortică supravalvulară (SASV)
II. Anomalii ale tractului de ieşire din ventriculul stâng b. Stenoza aortică supravalvulară (SASV) Definiţia SASV = afecţiune cu mecanism de transmitere AD caracterizată prin îngustarea congenitală a aortei ascendente ce cuprinde o zona variată de deasupra emergenţei arterelor coronare. Gena responsabilă se găseşte pe aceeaşi subunitate cu elastina, la nivelul braţului lung al cromozomului 7. Leziunea anatomică : hipoplazia aortei ascendente realizată pe seama dezorganizării mediei aortei.
 Definiţia. SASV = afecţiune cu mecanism de transmitere AD caracterizată prin îngustarea congenitală a aortei ascendente ce cuprinde o zona variată de deasupra emergenţei arterelor coronare. Gena responsabilă se găseşte pe aceeaşi subunitate cu elastina, la nivelul braţului lung al cromozomului 7. Leziunea anatomică : hipoplazia aortei ascendente realizată pe seama dezorganizării mediei aortei.")
83
b. Stenoza aortică supravalvulară (SASV)
II. Anomalii ale tractului de ieşire din ventriculul stâng b. Stenoza aortică supravalvulară (SASV) Definiţia SASV = afecţiune cu mecanism de transmitere AD caracterizată prin îngustarea congenitală a aortei ascendente ce cuprinde o zona variată de deasupra emergenţei arterelor coronare. Gena responsabilă se găseşte pe aceeaşi subunitate cu elastina, la nivelul braţului lung al cromozomului 7. Leziunea anatomică : hipoplazia aortei ascendente realizată pe seama dezorganizării mediei aortei.
 Definiţia. SASV = afecţiune cu mecanism de transmitere AD caracterizată prin îngustarea congenitală a aortei ascendente ce cuprinde o zona variată de deasupra emergenţei arterelor coronare. Gena responsabilă se găseşte pe aceeaşi subunitate cu elastina, la nivelul braţului lung al cromozomului 7. Leziunea anatomică : hipoplazia aortei ascendente realizată pe seama dezorganizării mediei aortei.")
84
singura alternativă terapeutică
Tabloul clinic Leziunea aortică poate fi izolată sau în cadrul tabloului clinic al sindromului Williams caracterizat prin: facies particular ("elfin"), hipercalcemie şi boală vasculară (SASV, leziuni coronariene, renale etc). Simptomatologia clinică seamănă cu cea din stenoza aortică valvulară putând dezvolta insuficienţă cardiacă chiar la sugari, infarct sau moarte subită. EKG : semne de HVS Tratament chirurgical: singura alternativă terapeutică constă din înlocuirea aortei hipoplastice cu grefa prostetică.
, hipercalcemie şi. boală vasculară (SASV, leziuni coronariene, renale etc). Simptomatologia clinică seamănă cu cea din stenoza aortică valvulară putând dezvolta insuficienţă cardiacă chiar la sugari, infarct sau moarte subită. EKG : semne de HVS. Tratament. chirurgical: singura alternativă terapeutică. constă din înlocuirea aortei hipoplastice cu grefa prostetică.")