Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
DEFICITELE IMUNE LA COPIL
Released by MedTorrents.com

2
Internat de urgenţă în perioada 30 V-16 VI 2000 MEDIU: urban
Caz clinic 1 B.S., 5 ani, sex masculin Internat de urgenţă în perioada 30 V-16 VI 2000 MEDIU: urban Diagnostic de trimitere: Sindrom febril prelungit M.I.: stare generală mediu alterată febră (39-400C) tuse seacă iritativă
 tuse seacă iritativă.")
3
AHC: APF: tata - 27 de ani, bronşite repetate
mama - 26 de ani, aritmie extrasistolică APF: primul copil, provenit din sarcină fiziologică, naştere spontană, născut la 9 luni, G=3000 gr, L=54cm., Apgar=10 alimentat natural până la 3 luni, apoi artificial (LP iar în continuare LV) vaccinat şi vitaminizat conform schemei MS (cicatrice BCG prezentă)
 vaccinat şi vitaminizat conform schemei MS (cicatrice BCG prezentă)")
4
Condiţii de viaţă: corespunzătoare
APP 2 săptămâni - larigintă acută de la 4 luni: infecţii respiratorii repetate (1-2/lună) reprezentate de: rinofaringite, laringite, bronşiolite, bronşite, pneumonii de la 2 ani: otite (1-2/lună) Condiţii de viaţă: corespunzătoare
 reprezentate de: rinofaringite, laringite, bronşiolite, bronşite, pneumonii. de la 2 ani: otite (1-2/lună) Condiţii de viaţă: corespunzătoare.")
5
IBA: Boala debutează brusc cu o săptămână anterior internării prin apariţia febrei C, care cedează greu la administrarea antitermicelor, alterarea stării generale. Se adresează medicului de familie care stabileşte diagnosticul de pneumonie acută si recomandă tratament cu Penicilină şi fluidificante ale secreţiei bronşice, sub care evoluţia este nefavorabilă. Se adresează serviciulul nostru pentru diagnostic şi tratament de specialitate.

6
Examen obiectiv la internare
stare generală uşor modificată febră 390C, G=15 kg ( deficit ponderal de 4 kg), T=99,8 cm (-5cm.), tegumente palide, uscate, prezenţa de lez. pruriginoase, ţesut celular subcutanat uşor diminuat, faringe discret congestionat, MV uşor înăsprit la nivelul bazei hemitoracelui drept, rare raluri crepitante la acest nivel, FR=28 resp/min.
, T=99,8 cm (-5cm.), tegumente palide, uscate, prezenţa de lez. pruriginoase, ţesut celular subcutanat uşor diminuat, faringe discret congestionat, MV uşor înăsprit la nivelul bazei hemitoracelui drept, rare raluri crepitante la acest nivel, FR=28 resp/min.")
7
Reţinem din datele anterioare:
băiat de 5 ani de la 2 săptămâni inf. resp. superioare şi inferioare frecvente alimentat natural 3 luni tatăl - bronsite repetate hipotrofia staturo-ponderală

8
INTERPRETARE CLINICA 1. Infecţii de căi respiratorii superioare şi inferioare repetate. 2. Deficit imun - inf. resp. repetate. 3. Teren atopic - lez. pruriginoase cutanate. 4. Sindrom anemic - prin mecanism patogenetic mixt (carenţial şi intrainfecţios). 5. Hipotrofie staturo - ponderală.
. 5. Hipotrofie staturo - ponderală.")
10
Radiografie toracică (30
Radiografie toracică (30.05): Multiple microopacităţi miliariforme diseminate paracardiac şi bazal bilateral, opacitate inomogenă cu bronhogramă aerică localizată la nivelul lobului mediu, dreapta, cu delimitare difuză spre parenchim. Cord normal. Focare micronodulare diseminate bilateral, hilar şi infrahilar, pe un fond de tramă intersţială accentuată difuz. Radiografie pumn stâng (12.06): Osteotransparenţă, vârstă osoasă la 2 1/2 ani. Ex ORL: Otita medie supurată dreaptă.
: Multiple microopacităţi miliariforme diseminate paracardiac şi bazal bilateral, opacitate inomogenă cu bronhogramă aerică localizată la nivelul lobului mediu, dreapta, cu delimitare difuză spre parenchim. Cord normal Focare micronodulare diseminate bilateral, hilar şi infrahilar, pe un fond de tramă intersţială accentuată difuz. Radiografie pumn stâng (12.06): Osteotransparenţă, vârstă osoasă la 2 1/2 ani. Ex ORL: Otita medie supurată dreaptă.")
11
DIAGNOSTIC POZITIV 1. Bronhopneumonie cu tendinţă la confluere, de etiologie neprecizată (datorită antibioterapiei de durată) cu IR gr. I. 2.Otită medie acută supurată. 3. Prurigo strophulus. 4. Anemie hipocromă intrainfecţioasă şi carenţială. 5. Hipotrofie staturo-ponderală. 6. Agamaglobulinemie.

12
Agamaglobulinemie, situaţie în care Ig totale < 100 mg%
AG. X linkată- Bruton AG. cu transmitere AR AGAMMAGLOBULINEMIE BRUTON Argumente: sex- masculin (transmiterea legata de cromosomul X) absenţa totală a Ig A; M; G; E Contraargumente prezenţa amigdalelor palatine şi a vegetaţiilor adenoide (adenoidectomizat) nu a prezentat infecţii grave cu germeni încapsulaţi piogeni (stafilococi, streptococi, HI, pneumococi) cu evoluţie spre septicemii, meningite, artrite. debut anterior vârstei de 4 luni (2 săpt.), imunitatea umorală fiind asigurată de Ig materne.
 absenţa totală a Ig A; M; G; E. Contraargumente. prezenţa amigdalelor palatine şi a vegetaţiilor adenoide (adenoidectomizat) nu a prezentat infecţii grave cu germeni încapsulaţi piogeni (stafilococi, streptococi, HI, pneumococi) cu evoluţie spre septicemii, meningite, artrite. debut anterior vârstei de 4 luni (2 săpt.), imunitatea umorală fiind asigurată de Ig materne.")
13
Agammaglobulinemie cu transmitere AR
Diagnostic cert: biopsia unui ganglion după stimulare antigenică, când nu se evidenţiază organizare foliculară şi absenţa plasmocitelor. cariotip, defectul genetic localizat pe Xq 21-22, la nivelul genei ce codifică proteina btk (împiedică maturarea şi diferenţierea celulelor B) Agammaglobulinemie cu transmitere AR paraclinic asemănătoare cu AG X clinic cu manifestări mai estompate tatăl cu bronşite repetate
 Agammaglobulinemie cu transmitere AR. paraclinic asemănătoare cu AG X. clinic cu manifestări mai estompate. tatăl cu bronşite repetate.")
14
PROBLEME DE DIAGNOSTIC
1. Ig administrate i.v., lunar, conţin predominant IgG, ceea ce determină valori >100 mg/mI. 2. Deficit calitativ de IgG cu N/hipogamaglobulinemie în proteinogramă (în clinică valori de 14%, 10%). 3. Deficit selectiv total de IgA asociat cu deficit de IgE, aceste cazuri evoluând cu manifestări pulmonare mai puţin grave, comparativ cu cele în care deficitul de IgA se asociază cu hiper IgE. În acest ultim caz sunt favorizate infecţiile şi manifestările alergice (pacientul dg. în antecedente cu astm bronşic), iar în timp prin administrarea de Ig i.v. poate apare şoc anafilactic.
. 3. Deficit selectiv total de IgA asociat cu deficit de IgE, aceste cazuri evoluând cu manifestări pulmonare mai puţin grave, comparativ cu cele în care deficitul de IgA se asociază cu hiper IgE. În acest ultim caz sunt favorizate infecţiile şi manifestările alergice (pacientul dg. în antecedente cu astm bronşic), iar în timp prin administrarea de Ig i.v. poate apare şoc anafilactic.")
15
4. Hipogamaglobulinemia X linkată cu deficit de STH
În acest caz este caracteristică: prezenţa L B în circulaţie, dar cu funcţie alterată sunt prezente amigdalele palatine nanism armonic

16
5. Hipogamaglobulinemia comună variabilă
din 1971 inclusă în imunodeficienţele primare, existând şi un mecanism genetic în determinismul bolii, pe lângă factorii infecţioşi virali specifici: Epstein Barr, rujeola, rubeola. Pacientul a prezentat o perioadă de 5 ani de infecţii repetate, dar nu cu virusurile menţionate. Clinic este asemănătoare cu AGX, însă în evoluţie prezintă infecţii mai puţin severe şi există o dezvoltare normală/hiperplazică a ţesutului limfoid (pacientul a fost adenoidectomizat). Paraclinic prezintă valori Ig totale < 350mg%, IgE < 250mg%; LB sunt prezente în circulaţie dar au funcţia alterată.
. Paraclinic prezintă valori Ig totale < 350mg%, IgE < 250mg%; LB sunt prezente în circulaţie dar au funcţia alterată.")
17
6. Alte deficite imune Celulare Mixte Ale complementului
Ale fagocitozel

18
TRATAM E NT 1. Substituţie cronică cu imunglobuline administrate: i.v mg/kg/doză la interval de 2-6 saptamani sau i.m. 0,2-0,6 ml/kg/lună din sol. de 16,5% astfel încât să menţinem IgG la valori de mg%. OCTAGAM 2,5 g/doză/lună 2. Imunomodulator: Zinc -1/2 tb. de 100 mg. 3. Antibiotic: Zinacef (50 mg/kg) apoi Rocephin (l00 mg/kg) asociat cu Amikacină (15 mg/kg) 4. Mucolitice: Fluimucil şi aerosoli cu Mucosolvin 5. Antimflamator: Nurofen
 apoi Rocephin (l00 mg/kg) asociat cu Amikacină (15 mg/kg) 4. Mucolitice: Fluimucil şi aerosoli cu Mucosolvin. 5. Antimflamator: Nurofen.")
19
Evoluţia Favorabilă clinic: stare generală bună, G=18 kg (-1kg), T=103 (-0,2cm) şi paraclinic cu reducerea sdr. inflamator. Dar pacientul prezintă la sfârşitul fiecărei luni un episod de IACRS şi otite, şi se practică adenoidectomie în luna IX 2000, urmată de o reducere marcată a sdr. inflamator (VSH=5-10).
, T=103 (-0,2cm) şi paraclinic cu reducerea sdr. inflamator. Dar pacientul prezintă la sfârşitul fiecărei luni un episod de IACRS şi otite, şi se practică adenoidectomie în luna IX 2000, urmată de o reducere marcată a sdr. inflamator (VSH=5-10).")
20
Rezervat datorită complicaţiior:
Prognostic Rezervat datorită complicaţiior: Bronşiectazii, deces în decada 4-a prin cord pulomar cronic Virajul spre o boală autoimună: LES, ARJ. lnfecţii generalizate determinând septicemii, cauză de deces la orice vârstă. 20-40 %-artrite 6% evoluează spre malignizare.

21
Particularitatea cazului
1. Precizarea tardivă a terenului care a favorizat patologia infecţioasă respiratorie, diagnosticul fiind pus la vârsta de 5 ani, cu ocazia primei internari în CI. Pediatrie II. 2. Evoluţia, pe acest fond de deficit imun total, fără a prezenta infecţii severe: septicemii, meningite, artrite. 3. Clinic se remarcă prezenţa amigdalelor palatine şi a vegetaţiilor adenoidiene, aspect ce contrazice diagnosticul de AGX şi pledează pentru AG cu transmitere AR.

23
Caz clinic 2 R.R., 4,5 luni, mediu urban Motivele internării:
Scaune diareice muco-grunjoase; Inapetenţă; Stagnare ponderală; Febră; Eczemă la nivelul feţei. AHC: - tata 30 ani, sănătos - mama 27 ani, sănătoasă - soră 3 ani, sănătoasă - neagă contact TBC sau alte suferinţe cronice APF: al doilea copil al mamei, provenit din a 2-a sarcină, fiziologică, născut la termen, G naştere 3400g, talia 50 cm, prezentaţie craniană, scor Apgar 7, icter fiziologic 4 zile, alimentat natural de la naştere până în momentul primei internări, ulterior până la vârsta de 6,5 luni, apoi artificial, diversificare la 3,5 luni, corect. Vaccinat BCG şi DTP, vitaminizat D (doze stos).
.")
24
rinofaringită, otită la 2,5 luni infecţie urinară la 3 luni (E.Coli)
APP: rinofaringită, otită la 2,5 luni infecţie urinară la 3 luni (E.Coli) Istoricul bolii: Debut insidios în urmă cu aprox. 2 luni prin inapetenţă, febră. Urmează tratament antibiotic ambulator (Penicilină G apoi Ampicilină) la indicaţia medicului de familie care a interpretat iniţial cazul ca Rinofaringită acută cu reacţie otică dreaptă. În ciuda acestui tratament efectuat timp de 7 zile simptomatologia persistă, în plus apar şi scaune diareice muco-grunjoase, inapetenţă, motiv pentru care se internează în clinică unde se stabileşte diagnosticul de Infecţie urinară cu E.Coli şi urmează tratament cu Colimicină + Gentamicină i.v. Timp de 10 zile, apoi se externează uşor ameliorat, dar cu persistenţa subfebrilităţilor, a scaunelor semiconsistente, a inapetenţei, simptomatologie care determină aparţinătorii să solicite internarea în clinica noastră.
 Istoricul bolii: Debut insidios în urmă cu aprox. 2 luni prin inapetenţă, febră. Urmează tratament antibiotic ambulator (Penicilină G apoi Ampicilină) la indicaţia medicului de familie care a interpretat iniţial cazul ca Rinofaringită acută cu reacţie otică dreaptă. În ciuda acestui tratament efectuat timp de 7 zile simptomatologia persistă, în plus apar şi scaune diareice muco-grunjoase, inapetenţă, motiv pentru care se internează în clinică unde se stabileşte diagnosticul de Infecţie urinară cu E.Coli şi urmează tratament cu Colimicină + Gentamicină i.v. Timp de 10 zile, apoi se externează uşor ameliorat, dar cu persistenţa subfebrilităţilor, a scaunelor semiconsistente, a inapetenţei, simptomatologie care determină aparţinătorii să solicite internarea în clinica noastră.")
25
STAREA LA INTERNARE: - Temperatura 37,70 C - Greutatea: 6400 g - Indice ponderal: 0,9 Stare generală satisfăcătoare, tegumente palide; Eczemă la nivelul obrajilor (prezentă din prima lună de viaţă); Eritem inghino-scrotal şi fesier; Ţesut celular subcutanat mai redus reprezentat pe abdomen, turgor normal; Ficat la aproximativ 2,5 cm sub rebord, marginea inferioară rotunjită, consistenţă normală.
; Eritem inghino-scrotal şi fesier; Ţesut celular subcutanat mai redus reprezentat pe abdomen, turgor normal; Ficat la aproximativ 2,5 cm sub rebord, marginea inferioară rotunjită, consistenţă normală.")
26
DIAGNOSTIC DE ETAPĂ: 1. Boală diareică trenantă (enterală şi/sau parenterală) de natură infecţioasă. 2. Suspect deficit imun. 3. Distrofie gradul I. 4. Anemie. 5. Eczemă.

27
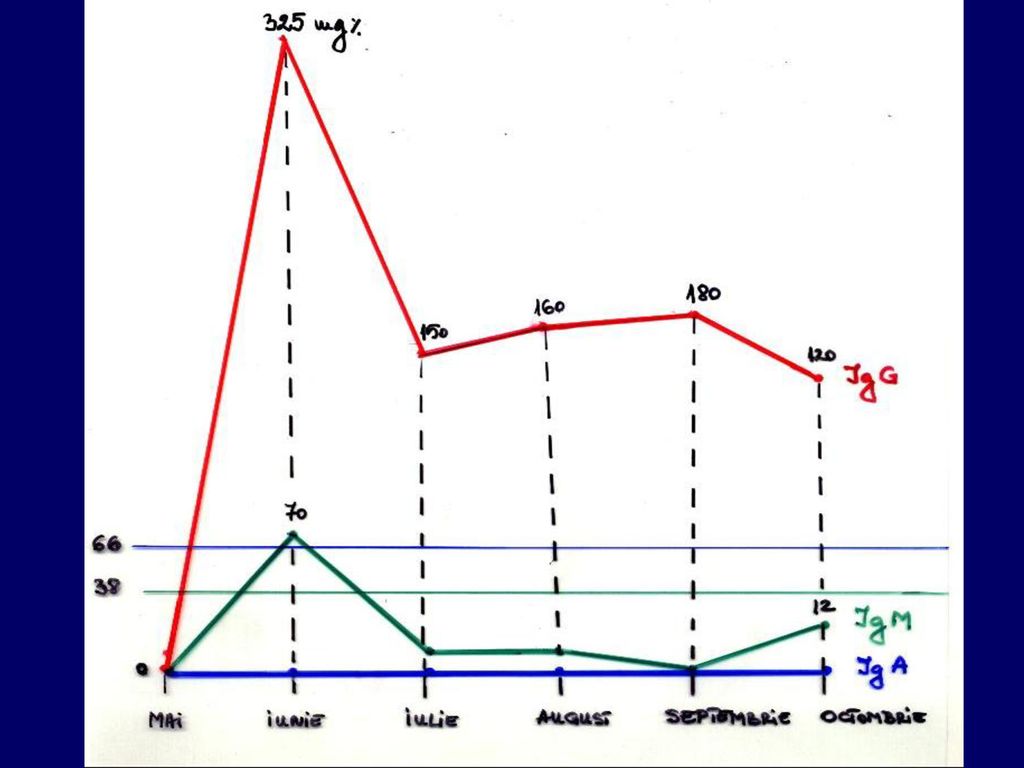
HEMATOLOGIE L= /mm3 Tablou sanguin: NNS= 2%, Mo=13% NS=15%, Eo=2% Ly=68% (Proporţie crescută de Ly mari, cu nucleu mare şi cromatină mai puţin condensată într-o reţea fină, citoplasmă bazofilă Tr = /mm3 Hb=9,5 g% Ht =36% H= /mm3 VSH=15-36 mm/h BIOCHIMIE 23.IX. CIC=20x10-3 UF C3=80 mg% IgG=14 UI/ml IgA=41 UI/ml IgM=20 UI/ml 4.X. IgG= 12 UI/ml IgA= 32 UI/ml IgM= 16 UI/ml ASAT=13 mU/ml ALAT= 10 mU/ml Tymol=1,1 uML Colinesteraza=3,8 U/ml BACTERIOLOGIE Coproculturi: I. E.Coli enteropatogen II. E.Coli Klebsiella Enterobacter Levuri Urocultură: sterilă Hemoculturi: sterile SF-Staf. aureu hemolitic E.Coli

28
Examen ORL: rinofaringită acută otic relaţii normale Radiografie toracică: absenţa radiologică a timusului Investigaţii virusologice: AgHBs - negativ Ac. faţă de virusurile gripale, paragripale, adenovirus, coronavirus, Coxsackie - negative R. Paul-Bunnel - negativă HIV - negativ Ac. faţă de CMV - în lucru pe perioada primei internări

29
Interpretare clinică:
1. Sindrom Wiskott-Aldrich enterocolita trenantă cu gram negativi (EPEC) eczema APP: infecţie urinară, rinofaringite, otită ? valorile scăzute ale IgM şi IgG absenţa radiologică a timusului 2. Distrofie gr. I 3. Anemie carenţială şi intrainfecţioasă
 eczema. APP: infecţie urinară, rinofaringite, otită valorile scăzute ale IgM şi IgG. absenţa radiologică a timusului. 2. Distrofie gr. I. 3. Anemie carenţială şi intrainfecţioasă.")
30
SINDROMUL WISKOTT-ALDRICH
- Este o boală ereditară cu transmitere recesivă legată de sex (doar sexul masculin prezintă boala, sexul feminin doar o transmite). - Face parte din grupul bolilor imunodeficitare combinate (atât deficit imun umoral cât şi celular). - Debut de regulă din primele luni de viaţă. Tabloul clinic: - infecţii recurente, trenante, digestive, respiratorii, ORL, urinare etc., cu etiologie bacteriană, virală, fungică, protozoare; - eczemă; - trombocitopenie cu manifestări hemoragice.
. - Face parte din grupul bolilor imunodeficitare combinate (atât deficit imun umoral cât şi celular). - Debut de regulă din primele luni de viaţă. Tabloul clinic: - infecţii recurente, trenante, digestive, respiratorii, ORL, urinare etc., cu etiologie bacteriană, virală, fungică, protozoare; - eczemă; - trombocitopenie cu manifestări hemoragice.")
31
Imuno-biologic: Deficit imun celular - timus displazic sau hipoplazic - depleţia ariilor paracorticale timodependente din ganglionii limfatici - reacţiile de hipersensibilitate, rejecţia allografelor şi TTL la PHA - alterate Deficit imun umoral - scăderea IgM + IgG - răspuns slab la Ag bacteriene polizaharidice - uneori scăderea IgA serice şi secretorii şi a IgG

32
Tratament Gammaglobulină i.v.; antibioterapie în asociaţie (Zinacef + Colimicină, apoi Fortum); eubiotice intestinale; tratament cu unguent cortizonic la nivelul feţei (pt. eczeme), băi de muşeţel şi amidon; măsuri dietetice adecvate evoluţiei enterocolitei.
, băi de muşeţel şi amidon; măsuri dietetice adecvate evoluţiei enterocolitei.")
33
Evoluţia (în decurs de o lună de spitalizare) a fost lent favorabilă cu ameliorarea progresivă a aspectului scaunelor, negativarea coproculturilor, ameliorarea aspectului dermatitei eczematiforme (fără însă ca ea să dispară), revenirea apetitului şi reluarea lentă a creşterii ponderale, dar cu persistenţa subfebrilităţilor. După aproximativ o lună revine în clinică pentru tuse, dispnee, febră, alterarea progresivă a stării generale, pe fondul persistenţei scaunelor muco-grunjoase, a inapetenţei, a stagnării ponderale şi a extinderii eczemei în paralel cu apariţia de peteşii.

34
Examenul obiectiv la această a doua internare a evidenţiat stare generală alterată, tuse seacă iritativă, dispnee moderată cu polipnee ~ 40 resp./min., tiraj moderat inter- şi subcostal, murmur vezicular înăsprit, raluri crepitante şi subcrepitante diseminate, iniţial fără semne de decompensare cardiacă, eczemă extinsă la nivelul feţei şi a regiunii inghinale şi fesiere cu descuamaţie, purpură tegumentară la nivelul membrelor, hepatomegalie la aproximativ 2 cm sub rebord cu marginea rotunjită, splina palpabilă la circa 3 cm sub rebord, stare de nutriţie deficitară (G=5800 g), anemie. Examenul radiologic a relevat un aspect de bronhopneumonie şi absenţa timusului.

35
Investigaţiile de laborator au arătat:
- trombocitopenie ~ /mm3 - leucocitoză /mm3 cu neutrofilie (68%) în tabloul sanguin - VSH accelerată mm/h Examinările bacteriologice au relevat prezenţa în secreţia faringiană şi spută de Stafilococ aureu hemolitic, E. Coli şi Enterobacter, în coproculturi de E.Coli, Klebsiella şi Enterobacter, iar în urocultură de E.Coli. Hemoculturi au fost sterile. S-au repetat dozările imunologice: IgG 22 UI/ml, IgA 42 UI/ml, IgM 18 UI/ml. Investigaţiile virusologice (al căror rezultat ne-a parvenit tardiv) au arătat prezenţa de anticorpi anticitomegalovirus de tip IgM.
 în tabloul sanguin. - VSH accelerată mm/h. Examinările bacteriologice au relevat prezenţa în secreţia faringiană şi spută de Stafilococ aureu hemolitic, E. Coli şi Enterobacter, în coproculturi de E.Coli, Klebsiella şi Enterobacter, iar în urocultură de E.Coli. Hemoculturi au fost sterile. S-au repetat dozările imunologice: IgG 22 UI/ml, IgA 42 UI/ml, IgM 18 UI/ml. Investigaţiile virusologice (al căror rezultat ne-a parvenit tardiv) au arătat prezenţa de anticorpi anticitomegalovirus de tip IgM.")
36
S-a instituit tratament cu
Gammaglobulină i.v.; antibioterapie în asociaţie (Cloforan + Gentamicină); oxigenoterapie; simptomatice. Evoluţia a fost iniţial lent fevorabilă pentru ca apoi, pe un fond staţionar, să survină semnele clinico-radiologice ale unei miocardite acute care, în ciuda tratamentului, a evoluat nefavorabil determinând decesul.
; oxigenoterapie; simptomatice. Evoluţia a fost iniţial lent fevorabilă pentru ca apoi, pe un fond staţionar, să survină semnele clinico-radiologice ale unei miocardite acute care, în ciuda tratamentului, a evoluat nefavorabil determinând decesul.")
37
Caz clinic 3 Ţ.A., de sex masculin, 15 ani.
Internat de urgenţă în repetate rânduri în Clinica Pediatrie II . M I: dispnee, tuse chintoasă cu expectoraţie dificilă de spută purulentă, hipertermie (39,70C), alterarea profundă a stării generale pe fondul bolii de bază. AHC: nu există în familie cazuri care să prezinte boala de bază a pacientului (sindrom Louis-Bar). APF: fără importanţă pentru boala de bază.
, alterarea profundă a stării generale pe fondul bolii de bază. AHC: nu există în familie cazuri care să prezinte boala de bază a pacientului (sindrom Louis-Bar). APF: fără importanţă pentru boala de bază.")
38
APP: repetate bronhopneumopatii cu un episod de hemoptizie la vârsta de 10 ani, infecţii ORL (otite supurate, sinuzite, rinofaringite), infecţii urinare, care au impus spitalizări repetate în clinicile de Pediatrie, Boli Infecţioase, Pneumoftiziologie, ORL. Hepatită cronică cu AgHbs pozitiv (diagnosticată la vârsta de 6 ani) Dezvoltarea psiho-motorie şi şcolarizarea: retard psihic moderat dar cu caracter progresiv asociat cu disartrie şi infirmitate motorie severă evolutivă obligând la imobilizare la pat şi întreruperea şcolarizării în urmă cu aproximativ 2 ani.
, infecţii urinare, care au impus spitalizări repetate în clinicile de Pediatrie, Boli Infecţioase, Pneumoftiziologie, ORL. Hepatită cronică cu AgHbs pozitiv (diagnosticată la vârsta de 6 ani) Dezvoltarea psiho-motorie şi şcolarizarea: retard psihic moderat dar cu caracter progresiv asociat cu disartrie şi infirmitate motorie severă evolutivă obligând la imobilizare la pat şi întreruperea şcolarizării în urmă cu aproximativ 2 ani.")
39
Istoricul bolii: Boala de fond a debutat clinic manifest în jurul vârstei de 3 ani prin apariţia ataxiei cerebeloase statice şi dinamice cu dificultăţi în menţinerea ortostaţiunii (bază mai largă de susţinere, deplasări laterale cu tendinţă la cădere, şi a mersului ebrios, nesigur şi ulterior imobilizare), la care s-au asociat tremurăturile intenţionale şi miocloniile . În paralel copilul începe să prezinte numeroase episoade infecţioase bronhopulmonare şi otice de etiologie predominant bacteriană (pneumococ, H.Influenzae) cu evoluţie trenantă şi răspuns parţial la tratamentul antiinfecţios care au condus la instalarea progresivă a unei pneumopatii cronice fibrozante, a bronşiectaziei şi alterarea dezvoltării pondero-staturale.
, la care s-au asociat tremurăturile intenţionale şi miocloniile . În paralel copilul începe să prezinte numeroase episoade infecţioase bronhopulmonare şi otice de etiologie predominant bacteriană (pneumococ, H.Influenzae) cu evoluţie trenantă şi răspuns parţial la tratamentul antiinfecţios care au condus la instalarea progresivă a unei pneumopatii cronice fibrozante, a bronşiectaziei şi alterarea dezvoltării pondero-staturale.")
40
Afirmativ în jurul vârstei de 4 ani părinţii remarcă apariţia teleangiectaziilor la nivelul conjunctivelor bulbare, simetrice, roşii strălucitoare situate în regiunea mediană a globilor oculari. Mentinerea unei dezvoltări psihice normale a permis (în ciuda dificultăţilor motorii lent progresive) încadrarea în colectivitate (grădiniţă şi ulterior şcoală) chiar dacă repetatele intercurenţe infecţioase au impus spitalizări periodice. Cu ocazia acestora simptomatologia neurologică a cazului a fost interpretată în prima fază ca fiind în cadrul unei encefalopatii cronice infantile de etiologie neprecizată, acest termen generic nefiind satisfăcător în condiţiile în care bolnavul prezenta un tablou clinic mai complex (deficitul în IgA secretor fusese evidenţiat, iar teleangiectaziile au fost interpretate ca fiind o conjunctivită cronică bilaterală) iar în antecedente nu a existat un factor etiologic identificat care să determine degradarea motorie progresivă de tip cerebelos şi extrapiramidal.
 încadrarea în colectivitate (grădiniţă şi ulterior şcoală) chiar dacă repetatele intercurenţe infecţioase au impus spitalizări periodice. Cu ocazia acestora simptomatologia neurologică a cazului a fost interpretată în prima fază ca fiind în cadrul unei encefalopatii cronice infantile de etiologie neprecizată, acest termen generic nefiind satisfăcător în condiţiile în care bolnavul prezenta un tablou clinic mai complex (deficitul în IgA secretor fusese evidenţiat, iar teleangiectaziile au fost interpretate ca fiind o conjunctivită cronică bilaterală) iar în antecedente nu a existat un factor etiologic identificat care să determine degradarea motorie progresivă de tip cerebelos şi extrapiramidal.")
41
Persistenţa şi accentuarea triadei ataxie cerebeloasă, sindrom extrapiramidal + infecţii bronhopulmonare şi ORL pe fond de deficit total în IgA + teleangiectazii conjunctivale simetrice au obligat la reevaluarea cazului în clinica noastră şi stabilirea diagnosticului bolii de bază (sindrom Louis- Bar ) . În Clinica Pediatrie II Cluj-Napoca a fost internat în repetate rânduri pentru o simptomatologie similară cu cea prezentată (dispnee, tuse productivă cu expectoraţie purulentă, semne de insuficienţă cardiacă) survenită în context infecţios (febră, inapetenţă, alterarea stării generale), în acest fel fiind posibilă urmărirea evoluţiei afecţiunii ereditare.
 survenită în context infecţios (febră, inapetenţă, alterarea stării generale), în acest fel fiind posibilă urmărirea evoluţiei afecţiunii ereditare.")
42
Examenul obiectiv la internare: stare generală profund alterată, adinamie, anxietate, tuse epuizantă prin efortul de expectoraţie, dispnee. Facies imobil, aproape inexpresiv (“de mască”) cu surâs stereotip intermitent. Teleangiectazii simetrice, roşii strălucitoare, la nivelul conjunctivelor bulbare ocupând regiunile ecuatoriale ale ochilor şi oprindu-se la marginea limbului sclero-corneean. Teleangiectazii pe conjunctivele palpebrale. Tegumente şi mucoase palide. Stare de nutriţie deficitară (minus ponderal de 13 kg) cu ţesut subcutanat aproape dispărut, masă musculară hipotrofică mai ales la membrele inferioare. Aspectul copilului era aproape caşectic. Tonus muscular uşor crescut la nivelul membrelor inferioare. Poziţie particulară (distonică) determinată de cifoscolioză dorsală şi semiflexia membrelor inferioare.
 cu surâs stereotip intermitent. Teleangiectazii simetrice, roşii strălucitoare, la nivelul conjunctivelor bulbare ocupând regiunile ecuatoriale ale ochilor şi oprindu-se la marginea limbului sclero-corneean. Teleangiectazii pe conjunctivele palpebrale. Tegumente şi mucoase palide. Stare de nutriţie deficitară (minus ponderal de 13 kg) cu ţesut subcutanat aproape dispărut, masă musculară hipotrofică mai ales la membrele inferioare. Aspectul copilului era aproape caşectic. Tonus muscular uşor crescut la nivelul membrelor inferioare. Poziţie particulară (distonică) determinată de cifoscolioză dorsală şi semiflexia membrelor inferioare..")
43
Sistemul ganglionar periferic :nepalpabil (în ciuda repetatelor infecţii). Faringe uşor congestionat, amigdale atrofiate. Dispnee mixtă cu polipnee ~ 30 respiraţii / minut, moderat tiraj inter şi subcostal, uşoară cianoză periorală şi a extremităţilor. Degete hipocratice. Murmur vezicular înăsprit in ½ superioară a ambelor hemitorace, diminuat la baze, raluri bronşice, crepitante şi subcrepitante diseminate bilateral. Zgomote cardiace ritmice, frecvenţa 150/minut, de intensitate uşor crescută, fără sufluri. Ficat la 1 cm sub rebord, consistenţă de organ. Splina la rebord. Abdomen liber fără sensibilitate la palpare. Organe genitale externe hipoplazice, asimetrie a testiculilor (testiculul stâng mai mic).
..")
44
Sistemul nervos: stare de conştienţă prezentă
Sistemul nervos: stare de conştienţă prezentă. Reflexele osteo-tendinoase uşor diminuate, restul reflexelor nemodificate semnificativ. Ataxie statică cu tremurături, hipermetrie, dismetrie, tremurătură intenţională, adiadocokinezie. Imposibilitatea ortostaţiunii şi implicit a mersului. Prezintă disartrie cu vorbirea lentă, sacadată. Dezvoltarea psihică retardată la nivelul unei întârzieri mintale uşoare/medii. Apraxie oculară. Asinergie oculo-cefalică Investigaţiile de laborator au evidenţiat caracteristicile unei infecţii bacteriene (leucocitoză cu neutrofilie, VSH accelerată, fibrinogen crescut, proteina C reactivă pozitivă), confirmată de prezenţa în exudatul faringian şi spută de S. Pneumoniae, Acinetobacter şi Candida, pe fondul unui deficit total în IgA, hipergamaglobulinemie, hipoxemie (saturaţia în oxigen 68%). Anergie tuberculinică.
, confirmată de prezenţa în exudatul faringian şi spută de S. Pneumoniae, Acinetobacter şi Candida, pe fondul unui deficit total în IgA, hipergamaglobulinemie, hipoxemie (saturaţia în oxigen 68%). Anergie tuberculinică.")
45
Examenul radiologic pulmonar (în dinamică) a evidenţiat un aspect de fibroză pulmonară interstiţială difuză, bronşiectazie şi un cord mic. Examenul ECG a relevat o deviere a axei QRS la +900 şi un interval QT prelungit. Explorare funcţională pulmonară : obstrucţie medie pe căile aeriene periferice. Severă hiperinflaţie pulmonară. Rezervele ventilatorii reduse 44%.

46
Pe baza acestor date am stabilit următorul diagnostic:
1. ATAXIE- TELEANGIECTAZIE ( SINDROM LOUIS-BAR): ataxie cerebeloasă + teleangiectazii conjunctivale + infecţii respiratorii, ORL severe, recidivante pe fond de deficit imun (IgA serice absente) + sindrom extrapiramidal + tulburările motricităţii oculare. 2. Bronhopneumopatie cronică acutizată cu insuficienţă cardio-respiratorie gradul II. Fibroză pulmonară. Bronşiectazie. 3. Hepatită cronică B. 4. Hipotrofie ponderală severă (caşeczie). 5. Anemie carenţială şi intrainfecţioasă.
: ataxie cerebeloasă + teleangiectazii conjunctivale + infecţii respiratorii, ORL severe, recidivante pe fond de deficit imun (IgA serice absente) + sindrom extrapiramidal + tulburările motricităţii oculare. 2. Bronhopneumopatie cronică acutizată cu insuficienţă cardio-respiratorie gradul II. Fibroză pulmonară. Bronşiectazie. 3. Hepatită cronică B. 4. Hipotrofie ponderală severă (caşeczie). 5. Anemie carenţială şi intrainfecţioasă.")
47
Tratament: antibioterapie în asociaţie (ex. Fortum+Amikacină), gammaglobulina intravenos, mucolitice, aerosoloterapie, fizioterapie, tratament tonic cardiac şi diuretic, oxigenoterapie, vitaminoterapie, alimentaţie hipercalorică, hiperproteică, tratamentul ataxiei (Majeptil), psihoterapie, evitarea contactelor infecţioase Evoluţia şi prognosticul au fost de la început rezervate având în vedere imobilizarea la pat a bolnavului, frecventele episoade infecţioase care s-au suprapus peste o bronhopneumopatie cronică severă, un asemenea episod determinând decesul pacientului.
, psihoterapie, evitarea contactelor infecţioase. Evoluţia şi prognosticul au fost de la început rezervate având în vedere imobilizarea la pat a bolnavului, frecventele episoade infecţioase care s-au suprapus peste o bronhopneumopatie cronică severă, un asemenea episod determinând decesul pacientului.")
48
Caz clinic 4 T. A., 8 ani, mediu urban MI: este internat în clinica noastră, prin transfer de la Spitalul Sighet, diagnosticul de ieşire din această unitate medicală fiind de Stare septică, Anemie, Lambliază, pentru diagnostic şi tratament de specialitate. Starea la internare: G=17 kg (-10 kg), febra 380C, stare generală profund alterată, edeme palpebrale, herpes labial, tegumente palide, panariţiu police stâng, ţesut celular subcutanat slab reprezentat, edeme maleolare, adenopatie submandibulară laterocervicală, axială, inghinală, ganglioni mobili, nedureroşi, hipotonie, hipokinezie, murmur vezicular înăsprit, raluri crepitante şi subcrepitante bilateral, zgomote cardiace estompate, ficat la 4 cm sub rebord, splina la 10 cm sub rebord.
, febra 380C, stare generală profund alterată, edeme palpebrale, herpes labial, tegumente palide, panariţiu police stâng, ţesut celular subcutanat slab reprezentat, edeme maleolare, adenopatie submandibulară laterocervicală, axială, inghinală, ganglioni mobili, nedureroşi, hipotonie, hipokinezie, murmur vezicular înăsprit, raluri crepitante şi subcrepitante bilateral, zgomote cardiace estompate, ficat la 4 cm sub rebord, splina la 10 cm sub rebord.")
49
Diagnosticul de etapă a fost de:
Bronhopneumonie Miocardită cu insuficienţă cardiacă gr.II Sindrom spleno-adeno-hepatomegalic Hipotrofie ponderală Panarţiu. Herpes. Anemie.

50
EXAMINĂRI PARACLINICE
27.VIII.1997 L=3.000/mm3 Tr=60.000/mm3 H=2,6 mil/mm3 Hb=6,9 g% VSH=32-54 Ret=9 %o T.sang.: Ns=26% S=40% Ly=28% Mo=5% PMN cu granulaţii toxice Fier = 23,4 g% Fibrinogen=100 mg% Hemoculturi sterile Spută Micrococ Candida Ex.urină A: neg. Sed: 8-10 leucocite/c Rar cilindri hialini IDR negativ ASAT=34 mU/ml ALAT=17 mU/ml Colinesteraza=2,8 U/ml Uree=33,7 mg% Creatinina=0,85 mg% Bilirubina T, D, I = normală

51
29.VIIl.1997 L=4400 Tr=60.000/mmc Hb=7,2 g% Tr=65.000 Fibrinogen=80 mg% Na=133 mEq/ml K=3,5 mEq/ml Cl=88 mEq/ml Mg=2,12 mEq/ml P=4,2 mg% CIC=108x10-3 UF C3=12 mg% IgG=174 ui/ml IgA=158 ui/ml IgM=174 ui/ml Elisa: HIV pozitiv AgHBs pozitiv AcHBc pozitiv AcHBe poziv Examinările privind cordul relevă o miocardita cu bloc AV gr.I.

52
PUNCŢIE MEDULARĂ Maduvă cu celularitate bogată, raportul G/E uşor modificat în favoarea granulocitelor. Seria granulocitară, bogată, cu elemente în toate stadiile de maturaţie, deviată la stânga cu prezenţa anomaliei Pelger-Huet şi granulaţii toxice Seria eritrocitară prezentă cu elemente în toate stadiile de maturaţie, se observă un uşor grad de diseritropoieză (eritroblaşti oxifili cu nucleu deformat în treflă, polilobat), de asemenea eritroblaşti oxifili şi policromatofili cu citoplasma puţină şi zdrenţuită; unii eritroblaşti sunt megaloblastoizi şi prezintă usor asincronism de maturaţie nucleu-citoplasmă. Rari eritroblaşti bazofili binucleari. Seria limfoplasmocitară prezentă, ceva mai săracă. Megacariocite în toate stadiile de maturaţie, dar mai puţine trombocitogene. Particularitate: se pun in evidenţă celule reticulare atipice care nu se pot încadra.
, de asemenea eritroblaşti oxifili şi policromatofili cu citoplasma puţină şi zdrenţuită; unii eritroblaşti sunt megaloblastoizi şi prezintă usor asincronism de maturaţie nucleu-citoplasmă. Rari eritroblaşti bazofili binucleari. Seria limfoplasmocitară prezentă, ceva mai săracă. Megacariocite în toate stadiile de maturaţie, dar mai puţine trombocitogene. Particularitate: se pun in evidenţă celule reticulare atipice care nu se pot încadra.")
53
Diagnostic: Stare septică cu CID Bronhopneumonie
Miocardită cu insuficienţă cardiacă gr.ll Sindrom spleno-adeno-megalic Hepatită cronică B Pancitopenie prin: CID? HIV? Hipersplenism Panariţiu police stâng Herpes labial Hipofrofie staturo-ponderala SIDA (2 teste Elisa)
")
54
Investigarea HIV-ului a permis retroactiv diagnosticul de SIDA.
A urmat tratament cu: Antibiotice în dublă asociere Rochepin şi Gentamicină i.v. HHC i.v. Gammaglobulină i.v. Hepatoprotectoare Vasculotrope Tonicardiac Evoluţia a fost staţionară, cu uşoara ameliorare după 2-3 zile. S-a efectuat o transfuzie cu 150 ml sânge integral, fiind prezent CID-ul. În ziua de 31.VIII 1997 prezintă stare generală alterată, lipotimie, iar la orele 1230 prezintă stop cardiac ireversibil. Investigarea HIV-ului a permis retroactiv diagnosticul de SIDA. Examenul anatomopatologic a permis precizarea diagnosticului de Septicemie cu Pneumocytis Carini.

55
Deficitele imune primare
A. Clasificare I. Bolile primare ale celulelor B 1) Agammaglobulinemia X-linkată (XLA sau boala Bruton) 1: 2) Imunodeficienţa comună variabilă (ambele sexe !) 3) Deficitul selectiv în IgA (1:333 1:16.000) 4) Hipogammaglobulinemia tranzitorie a sugarului 5) Deficitul în subclase de IgG 6) Boala lanţurilor grele (absenţa totală de IgG1, IgG2, IgG4 şi/sau IgA1) 7) Boala limfoproliferativă X-linkată (răspuns inadecvat la infecţia cu virus Epstein-Barr)
 Agammaglobulinemia X-linkată (XLA sau boala Bruton) 1: ) Imunodeficienţa comună variabilă (ambele sexe !) 3) Deficitul selectiv în IgA (1:333 1:16.000) 4) Hipogammaglobulinemia tranzitorie a sugarului. 5) Deficitul în subclase de IgG. 6) Boala lanţurilor grele (absenţa totală de IgG1, IgG2, IgG4 şi/sau IgA1) 7) Boala limfoproliferativă X-linkată (răspuns inadecvat la infecţia cu virus Epstein-Barr)")
56
II. Bolile primare ale celulelor T
1) Hipoplazia timică (Sindromul DiGeorge) 2) Imunodeficienţa X-linkată (IgG + IgA ) cu hiper IgM 3) Expresie “defectuoasă” a receptorilor celulelor T - complexului CD3 (Ti-CD3) 4) Producţia “defectuoasă” de citokine 5) Limfocitopenia CD8 6) Defecte ale activării celulelor T
 Hipoplazia timică (Sindromul DiGeorge) 2) Imunodeficienţa X-linkată (IgG + IgA ) cu hiper IgM. 3) Expresie defectuoasă a receptorilor celulelor T - complexului CD3 (Ti-CD3) 4) Producţia defectuoasă de citokine. 5) Limfocitopenia CD8. 6) Defecte ale activării celulelor T.")
57
III. Bolile cu deficit combinat al celulelor B+T
1) Imunodeficienţa combinată (Sindromul Nezelof) 2) Deficitul în Purin-Nucleozid-Fosforilaza 3) Hipoplazia cartilaje-păr 4) Imunodeficienţa combinată severă (+ deficit al neutrofilului) 1: 1: ; X linkat 5) Imunodeficienţa combinată severă autosomal recesivă 6) Disgeneziile reticulare 7) Expresia defectuoasă a antigenelor complexului major de histocompatibilitate
 Imunodeficienţa combinată (Sindromul Nezelof) 2) Deficitul în Purin-Nucleozid-Fosforilaza. 3) Hipoplazia cartilaje-păr. 4) Imunodeficienţa combinată severă (+ deficit al neutrofilului) 1: 1: ; X linkat. 5) Imunodeficienţa combinată severă autosomal recesivă. 6) Disgeneziile reticulare. 7) Expresia defectuoasă a antigenelor complexului major de histocompatibilitate.")
58
8) Sindromul Omenn (imunodeficienţa combinată + hipereozinofilie); autosomal recesiv:
infecţii foarte grave infiltrarea cu celule T ale pielii, intestinului, ficatului, splinei; eritrodermie exfoliativă; limfadenopatie hepato-splenomegalie diaree intractabilă 9) Imunodeficienţa cu trombocitopenie şi eczemă (Sindrom Wiskott-Aldrich) 10) Sindromul hiperimunglobulinemie E (Job) 11) Ataxia - teleangectazia
 Imunodeficienţa cu trombocitopenie şi eczemă (Sindrom Wiskott-Aldrich) 10) Sindromul hiperimunglobulinemie E (Job) 11) Ataxia - teleangectazia.")
59
Deficitele imunitare la copil (abordare practică)
Patru mari sisteme participă, asociat, la răspunsul imunitar. În mod izolat sau combinat, ele pot fi afectate de o anomalie, sursă de deficit imun. Este vorba de: imunitate celulară legată de limfocitele T; imunitate umorală legată de limfocitele B; imunitate non-specifică legată de celulele fagocitare; imunitate non-specifică legată de sistemul complement

60
Orientarea diagnostică a unui deficit imunologic în faţa unei infecţii (după A. Fischer)
Pneumopatie interstiţială Diaree cronică Deficit profund al imunităţii Muguet bucal recidivant celulare Infecţii ORL + bronşice recidivante + Hipogammaglobulinemie diaree (lamblaiză) bronşice recidivante cu Deficit în IgG2 Haemophilus, Pseudomonas
 bronşice recidivante cu Deficit în IgG2. Haemophilus, Pseudomonas.")
61
Meningite, artrite, septicemii - Deficit în IgM
- Deficit într-o fracţiune de complement - Asplenie - Agranulocitoză Adenite, infecţiicutanate bacteriene, abces visceral Abces cutanat sau pulmonar Sindromul Buckley fără semne inflamatorii (+ alergie) Granulomatoză septică cronică
 Granulomatoză septică cronică.")
62
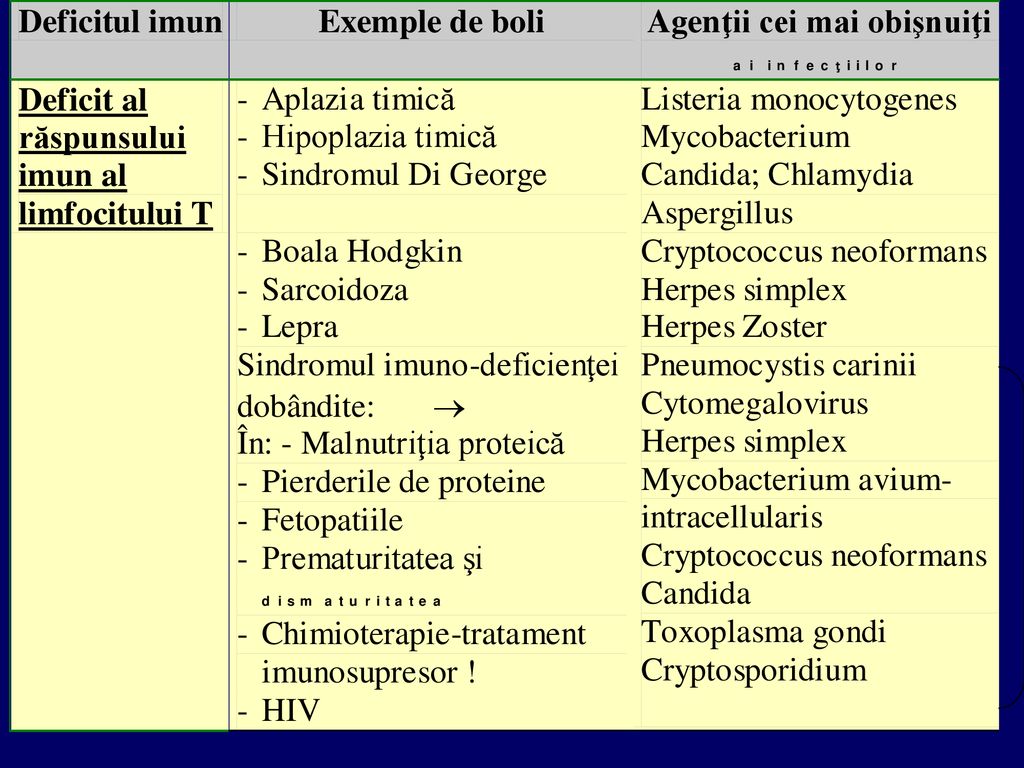
Infecţii asociate deficitelor imune (după Richard B. Roberts)
")
68
Deficitele imunităţii celulare
Limfocitele T produc limfokine capabile de a activa macrofagele pentru a distruge micro-organismele care “parazitează” obligatoriu sau facultativ aceste celule. Limfocitele T citotoxice distrug celulele infectate de către un virus, controlând astfel difuziunea virusului cu potenţial latent (virusurile grupului Herpes). În sfârşit, limfocitele T sunt necesare pentru producţie de anticorpi de către limfocitele B. Perturbarea acestor funcţii explică apariţia unor infecţii cu germeni zişi oportunişti.
. În sfârşit, limfocitele T sunt necesare pentru producţie de anticorpi de către limfocitele B. Perturbarea acestor funcţii explică apariţia unor infecţii cu germeni zişi oportunişti.")
69
La nivelul membranei LT mai există şi alţi markeri specifici:
Pe suprafaţa LT se disting 2 tipuri de receptori: TCR1 şi TCR2, care se asociază markerului major al LT, respectiv complexul TCR-CD3 (TCR1-CD3 şi TCR2-CD3 ). La nivelul membranei LT mai există şi alţi markeri specifici: CD4 care recunoaşte antigenul prezentat în asociere cu moleculele complexului major de histocompatibilitate de tip II (MHC tip II); CD8 care recunoaşte antigenul prezentat în asociere cu moleculele MHC de tip I.
. La nivelul membranei LT mai există şi alţi markeri specifici: CD4 care recunoaşte antigenul prezentat în asociere cu moleculele complexului major de histocompatibilitate de tip II (MHC tip II); CD8 care recunoaşte antigenul prezentat în asociere cu moleculele MHC de tip I.")
70
Limfocitele T care exprimă pe suprafaţa lor markeri TCR2-CD3 se împart în două tipuri:
limfocitele T helper (TH) care au markeri CD4+; limfocitele T citotoxice/supresoare (TC/S) care au markeri CD8+. La ora actuală se cunosc 5 subseturi de TH, în funcţie de tipul de limfokine pe care le sintetizează: THp, THo, TH1, TH2, THm. Limfocitele TH1 secretă: IL2, IFNγ şi limfotoxina. Limfocitele TH2 secretă: IL4, IL5, IL10.
 care au markeri CD4+; limfocitele T citotoxice/supresoare (TC/S) care au markeri CD8+. La ora actuală se cunosc 5 subseturi de TH, în funcţie de tipul de limfokine pe care le sintetizează: THp, THo, TH1, TH2, THm. Limfocitele TH1 secretă: IL2, IFNγ şi limfotoxina. Limfocitele TH2 secretă: IL4, IL5, IL10.")
71
Aceste limfocite LH1 şi LH2 provin din limfocitul CD4+, reprezentând o etapă finală în maturaţia acestor celule, secundar stimulării antigenice. Ambele clase de LH au capacitatea de a stimula limfocitul B (LB), dar această capacitate este net superioară pentru LH2. Limfocitele TH1 intervin în hipersensibilitatea de tip întârziat. Limfocitele TH2 mediază reacţia alergică, prin secreţia de IL4 care stimulează producţia de IgE şi IgG1. Ele favorizează reacţiile de hipersensibilitate imediată.
, dar această capacitate este net superioară pentru LH2. Limfocitele TH1 intervin în hipersensibilitatea de tip întârziat. Limfocitele TH2 mediază reacţia alergică, prin secreţia de IL4 care stimulează producţia de IgE şi IgG1. Ele favorizează reacţiile de hipersensibilitate imediată.")
72
Deficitele imunităţii umorale
Anticorpii recunosc antigenele din mediul extracelular. Ei neutralizează toxinele microbiene, inhibă fixarea virusurilor pe celulele lor ţintă, mai cu seamă fagocitează microorganismele recunoscute de celulele fagocitare. Ei sunt produşi în mod esenţial la nivelul mucoaselor (IgA şi IgG mai ales) şi participă la filtrul sanguin splenic. (IgM). De aceea, defectele de producţie de anticorpi produc infecţii bacteriene în special la nivelul mucoaselor. Deficitul în IgM provoacă un risc de septicemie şi de metastaze septice.
 şi participă la filtrul sanguin splenic. (IgM). De aceea, defectele de producţie de anticorpi produc infecţii bacteriene în special la nivelul mucoaselor. Deficitul în IgM provoacă un risc de septicemie şi de metastaze septice.")
73
Concetraţia serică (g/l) a IgG, A şi M la copii normali
 a IgG, A şi M la copii normali")
74
Simptomatologia deficitelor imune umorale

75
Tratament 1) Tratamentul prin imunoglobuline:
Tratamentul substitutiv cu Ig poate fi efectuat: pe cale i.v. folosind o Ig denumită intactă, cu o semidurată de viaţă apropiată de 21 zile, cu o repartiţie fiziologică a sub-claselor. produsele preparate la pH 4 în prezenţa unor urme de pepsină răspund acestei definiţii şi imunitatea lor faţă de transmisia HIV şi a hepatitei non A non B a fost perfect demonstrată.

76
- posologia obişnuită este de mg/kg la 3 săptămâni; dar ritmul şi posologia pot fi crescute în caz de infecţii sau de pierderi, de ex. pierderi pe cale digestivă a imunglobulinelor pe cale i.m. utilizează produşi 16% de origine plasmatică sau placentară în doză de 0,3-0,5 ml/kg la 2 săptămâni, este abandonată în deficitele imune umorale severe şi utilizarea sa este limitată în formele medii sau profilactic.

77
2) Tratamentul antibiotic:
Sunt posibile mai multe formule în funcţie de gravitatea deficitului imun umoral şi de tabloul clinic: Abţinerea terapeutică cu antibioterapie de necesitate cu un antibiotic bactericid, pentru o durată de minimum 10 zile. Monoantibioterapie cu Penicilină orală sau Ampicilină Antibioterapie alternativă bactericidă 20 zile /lună 3) Kinesiterapie respiratorie !!
 Kinesiterapie respiratorie !!")
78
4) Alte măsuri: Antifungice pe durată lungă; Tratament cu Metronidazol 5-10 zile/lună în caz de tulburări digestive, chiar dacă Lamblia nu a fost detectată; Tratament imunomodulator în caz de deficit în IgA (Biostin) Saliciloterapie şi macrolide în caz de “prindere” articulară; Vaccinări cu vaccinuri doar (numai) “omorâte” contra difteriei, tetanosului, poliomelitei, gripei, Pneumococului şi Haemofilului ori de cîte ori deficitul imun nu este complet.
 Saliciloterapie şi macrolide în caz de prindere articulară; Vaccinări cu vaccinuri doar (numai) omorâte contra difteriei, tetanosului, poliomelitei, gripei, Pneumococului şi Haemofilului ori de cîte ori deficitul imun nu este complet.")
79
DEFICITELE CELULELOR FAGOCITARE
Polinuclearele neutrofile migrează spre locul infectat, fagocitează bacteriile (sau ciupercile) cu dezvoltare extracelulară şi le “omoară”. Scăderea activităţii lor provoacă diseminarea infecţiiIor cu piogeni şi fungi. Neutropeniile copilului reprezintă o entitate cu etiologii multiple. Dacă, în ansamblu, neutropeniile tranzitorii prezintă un risc mai mic pentru copii, ţinand cont de progresele antibioterapiei, trebuie subliniate unele neutropenii cronice primitive pentru care noii hormoni hematopoetici deschid perspective terapeutice.
 cu dezvoltare extracelulară şi le omoară . Scăderea activităţii lor provoacă diseminarea infecţiiIor cu piogeni şi fungi. Neutropeniile copilului reprezintă o entitate cu etiologii multiple. Dacă, în ansamblu, neutropeniile tranzitorii prezintă un risc mai mic pentru copii, ţinand cont de progresele antibioterapiei, trebuie subliniate unele neutropenii cronice primitive pentru care noii hormoni hematopoetici deschid perspective terapeutice.")
80
CLASIFICAREA NEUTROPENIILOR LA COPIL
NEUTROPENIILE PRIMITIVE 1. Neutropenia cronică congenitală sau agranulocitoza congenitală 2.Neutropenia ciclică a copiluiui 3.Sindromul retenţiei medulare sau mielo-kathexis 4.Sindromul "lazy leucocyte" (chimiotactism anormal, în timp ce fagocitoza şi bactericidia PN sunt normale).
.")
81
NEUTROPENII ASOCIATE 1. Unor anomalii ale limfocitelor T şi B:
hipogammaglobulinemie (în special Ig G + IgA + IgM) (neutropenia se corijează după administrarea de gammaglobulină) hiperlimfocitoză 2. Unor boli ereditare: Sindromul Schwachmann (condrodisplazie metafizară + insuficienţă pancreatică exocrină + retard de creştere; medulograma poate arăta interesarea tuturor celor 3 linii) Sindromul Wiskott- Aldrich
 (neutropenia se corijează după administrarea de gammaglobulină) hiperlimfocitoză. 2. Unor boli ereditare: Sindromul Schwachmann (condrodisplazie metafizară + insuficienţă pancreatică exocrină + retard de creştere; medulograma poate arăta interesarea tuturor celor 3 linii) Sindromul Wiskott- Aldrich.")
82
3. Originii etnice : ex. aproximativ 30% din populaţia neagră prezintă o leucopenie inferioară la 5000/mm3 cu o neutropenie sub 300/mm3 4.Unor boli sistemice: LES ARJ Ciroza Hipersplenismul Gammopatiile monoclonale Hipertiroidismul B.Addison B. Crohn 5. Unor stări preleucemice (ex. LAM) 6. Unor boli înnăscute de metabolism: ex. glicogenoza tip Ib sau unele aminoacidopatii care evoluează cu hiperglicemie + cetoză.
 6. Unor boli înnăscute de metabolism: ex. glicogenoza tip Ib sau unele aminoacidopatii care evoluează cu hiperglicemie + cetoză.")
83
NEUTROPENII SECUNDARE
1.Unei infecţii: Infecţiile asociate cu neutropenie: Bacteriene: Febra tifoidă şi paratifoidă Tuberculoza Bruceloza Septicemia cu Gram negativi (forme grave)
")
84
- Virale: Hepatita lnfecţia cu VEB şi CMV Rujeola, rubeola, varicela, oreionul Infecţia cu v. gripale Poliomielita Febra galbenă Psittacoza - Infecţia cu Rickettsii: Febra “Munţiilor Rockenses” Tifosul - Micotice: Histoplasmosa - Parazitare: Paludium Leishmaniosa

85
2. Unor medicamente: Medicamentele responsabile de o neutropenie Antibacteriene: Carbenicilina Cefalosporine Cloramfenicol Penicilinele semisintetice Sulfamidele Vancomicina Antiinflamatoarele: Aminopirina Fenilbutazona

86
Antitiroidienele şi medicamentele antireumatismale:
Propil-thio-uracil Săruri de aur Hidroxicloroquina Tranchilizante: Clorpromazina Meprobamat Agenţi citostatici: Ciclofosfamida Methotrexat Cytosin-arabinozida 6 mercapto-purina Adriamicina

87
3. Unor carenţe nutriţionale (Vit B12, acid folic, cupru)
4. Unor toxine: chimioterapie iradiere intoxicaţie cu metale grele 5. Unei imunizări: neonatale (incompabilitate Rh) allo-imunizare (post multiple transfuzii) auto-imunizare izolată (neutropenia cronică benignă a copilului)
 allo-imunizare (post multiple transfuzii) auto-imunizare izolată (neutropenia cronică benignă a copilului)")
88
Diagnosticul deficitelor imune
Diagnosticul defictelor imune, suspectat în faţa unui tablou clinic sugestiv, poate fi susţinut de către diferite examinări de laborator: 1. Examinări de rutină: Hemoleucograma completă, în formula sanguină periferică interesându-ne numărul limfocitelor, monocitelor şi PMN; Electroforeza şi imunelectroforeza proteinelor serice.

89
2. Examinări imunitare în practica ambulatorie:
Aceste examinări explorează diferitele faze ale imunităţii: a) Studiul imunităţii celulare: Acestea sunt în mod esenţial reacţiile cutanate de hipersensibilitate întârziată: IDR la tuberculoză Testul la Candidină Multitestul IMC sau Multitest Mérieux Se poate asocia uneori testul rozetelor E b) Studiul imunităţii umorale: Dozarea cantitativă a Ig; Măsurarea nivelului anticorpilor, ceea ce permite aprecierea funcţiei lor.
 Studiul imunităţii celulare: Acestea sunt în mod esenţial reacţiile cutanate de hipersensibilitate întârziată: IDR la tuberculoză. Testul la Candidină. Multitestul IMC sau Multitest Mérieux. Se poate asocia uneori testul rozetelor E. b) Studiul imunităţii umorale: Dozarea cantitativă a Ig; Măsurarea nivelului anticorpilor, ceea ce permite aprecierea funcţiei lor.")
90
3. Examinări imunitare practicate în servicii specializate:
a) Explorarea imunităţii celulare: - Detectarea de sub-populaţii limfocitare prin anticorpi monoclonali: OKT4: limfocitele T helper sau facilitatoare OKT8: limfocitele T supresoare - Studiul aptitudinilor funcţionale ale limfocitului T: Testul de transformare limfoblastică Măsurarea funcţiilor de secreţie ale limfocitelor T (activitatea interferonului) Testul de migrare leucocitară
 Explorarea imunităţii celulare: - Detectarea de sub-populaţii limfocitare prin anticorpi monoclonali: OKT4: limfocitele T helper sau facilitatoare. OKT8: limfocitele T supresoare. - Studiul aptitudinilor funcţionale ale limfocitului T: Testul de transformare limfoblastică. Măsurarea funcţiilor de secreţie ale limfocitelor T (activitatea interferonului) Testul de migrare leucocitară.")
91
b) Explorarea imunităţii umorale:
Determinarea limfocitelor B care constituie 15-20% din limfocitele sanguine (se cere însă raportarea la vârstă !). Ele pot fi reperate datorită markerilor de suprafaţă situaţi pe membranele lor: prezenţa de Ig de suprafaţă (în special IgM) prezenţa de receptor Fc al IgG sau IgM prezenţa de receptor al fracţiunii C3 a complementului c) Explorarea celulelor fagocitare: Studiul chemotactismului Studiul endocitozei Studiul activităţii bactericide
. Ele pot fi reperate datorită markerilor de suprafaţă situaţi pe membranele lor: prezenţa de Ig de suprafaţă (în special IgM) prezenţa de receptor Fc al IgG sau IgM. prezenţa de receptor al fracţiunii C3 a complementului. c) Explorarea celulelor fagocitare: Studiul chemotactismului. Studiul endocitozei. Studiul activităţii bactericide.")
92
Subliniem că de mai mulţi ani:
datorită lavajului bronho-alveolar se pot studia diferitele populaţii celulare situate în arborele respirator datorită studiului mucusului salivar şi al mucusului bronşic prin intermediul tehnicilor de imun-fluorescenţă se pot doza IgAs. Pe lângă aceste explorări imunologice la care se adaugă cele bacteriologice, virusologice, fungice, este necesară dozarea serologică prin metode exacte pentru HIV în orice boală de etiologie trenantă, recidivantă sau cronică. Metodele de biotehnologie modernă trebuie neapărat introduse pentru depistarea etiologiei: Anticorpi monoclonali; Metode utilizând sondele de ADN

93
Relaţia DEFICIT IMUN - AUTOIMUNITATE - PATOLOGIE INFLAMATORIE CRONICĂ
Bolile autoimune (BAI) şi bolile inflamatorii ale ţesutului conjunctiv (BITC) sunt azi mai bine cunoscute în patologia infantilă (J.G.Schaller, M.J.McDuffie, 1996), insistându-se pe cascada reacţiilor imune ce caracterizează autoimunitatea (AI) şi pe terapia ţintită patogenetică. Termenul de AI semnifică răspunsul imunitar împotriva unor constituenţi proprii ai organismului. AI este un fenomen care aparţine fiziologiei sistemului imun, spre deosebire de BAI.
 şi bolile inflamatorii ale ţesutului conjunctiv (BITC) sunt azi mai bine cunoscute în patologia infantilă (J.G.Schaller, M.J.McDuffie, 1996), insistându-se pe cascada reacţiilor imune ce caracterizează autoimunitatea (AI) şi pe terapia ţintită patogenetică. Termenul de AI semnifică răspunsul imunitar împotriva unor constituenţi proprii ai organismului. AI este un fenomen care aparţine fiziologiei sistemului imun, spre deosebire de BAI.")
94
Există, deci, 2 fenomene distincte :
În timp ce AI constă din evenimente care se succed în mod normal, deci într-un organism sănătos, fiind un fenomen, deci, nepatogen, BAI reprezintă o parte importantă a patologiei actuale, atât la adult, cât şi la copil, ea fiind consecinţa unui proces autoimun care depăşeşte sfera fiziologicului, prin intensitatea şi durabilitatea sa. (J.Clot)
")
95
AUTOIMUNITATEA FIZIOLOGICĂ :
% din limfocitele B (LyB) normale pot produce autoanticorpi (AAc), polispecifici, capabili să reacţioneze cu mai mult de două antigene endogene diferite [Ac faţă de actină, tubulină, miozină, nucleu, tireoglobulină, IgG(FR)] şi chiar cu antigene exogene. Aceşti anticorpi au însă o constantă de asociere scăzută şi o afinitate slabă pentru autoantigene, sunt în principal de tip IgM, au spectru de recunoaştere destul de restrâns, găsindu-se într-un titru scăzut, dar având un rol în reglarea răspunsului imun. (J.M.Roitt, J.Brostoff, D.Male, 1994) Producţia acestor autoanticorpi în cantitate fiziologică este sub controlul limfocitelor T (LyT) , cu deosebire a LyT supresoare (LyTs), care frânează producţia lor, aceasta rămânând fiziologică.
 normale pot produce autoanticorpi (AAc), polispecifici, capabili să reacţioneze cu mai mult de două antigene endogene diferite [Ac faţă de actină, tubulină, miozină, nucleu, tireoglobulină, IgG(FR)] şi chiar cu antigene exogene. Aceşti anticorpi au însă o constantă de asociere scăzută şi o afinitate slabă pentru autoantigene, sunt în principal de tip IgM, au spectru de recunoaştere destul de restrâns, găsindu-se într-un titru scăzut, dar având un rol în reglarea răspunsului imun. (J.M.Roitt, J.Brostoff, D.Male, 1994) Producţia acestor autoanticorpi în cantitate fiziologică este sub controlul limfocitelor T (LyT) , cu deosebire a LyT supresoare (LyTs), care frânează producţia lor, aceasta rămânând fiziologică.")
96
BOALA AUTOIMUNĂ Ruptura toleranţei faţă de autoantigene conduce la instalarea bolii autoimune, cu producţie exagerată de autoanticorpi, cu sau fără specificitate de organ, în special prin intrarea în acţiune a LyT autoreactive (exces de LyT helper şi scăderea până la dispariţie a LyTs). Evantaiul bolilor autoimune este deosebit de mare :
. Evantaiul bolilor autoimune este deosebit de mare :")
97
Specificitate de organ
Tiroidita Hashimoto Mixedemul primar Boala lui Basedow Anemia pernicioasă (Biermer) Gastrita atrofică autoimună Boala lui Addison Diabetul zaharat insulino-dependent Sindromul Goodpasture Miastenia Sterilitatea masculină Pemfigusul vulgar Oftalmia simpatică Uveita facogenică Scleroza în plăci Anemia hemolitică autoimună Purpura trombocitopenică idiopatică Leucopenia idiopatică Ciroza biliară primitivă Hepatita cronică activă cu AgHBs-negativ Rectocolita hemoragică Sindromul Sjögren Poliartrita reumatoidă Dermatomiozita Sclerodermia Lupusul eritematos discoid Lupusul eritematos diseminat (LED) Fără specificitate de organ
 Gastrita atrofică autoimună. Boala lui Addison. Diabetul zaharat insulino-dependent. Sindromul Goodpasture. Miastenia. Sterilitatea masculină. Pemfigusul vulgar. Oftalmia simpatică. Uveita facogenică. Scleroza în plăci. Anemia hemolitică autoimună. Purpura trombocitopenică idiopatică. Leucopenia idiopatică. Ciroza biliară primitivă. Hepatita cronică activă cu AgHBs-negativ. Rectocolita hemoragică. Sindromul Sjögren. Poliartrita reumatoidă. Dermatomiozita. Sclerodermia. Lupusul eritematos discoid. Lupusul eritematos diseminat (LED) Fără specificitate de organ.")
98
Există boli caracterizate prin :
- specificitate de organ şi - cu expresie multiplă (LES) Asocierile între bolile situate la cele două extremităţi ale "evantaiului" sunt relativ puţin frecvente :
 Asocierile între bolile situate la cele două extremităţi ale evantaiului sunt relativ puţin frecvente :")
99
Clasificarea amintită nu ţine seama decât de mecanismele efectoare ultime ale bolii, şi nu de fenomenele declanşatoare. Unele boli autoimune sunt provocate de autoanticorpi şi sunt deci dependente de o imunitate B. Este cazul anemiei hemolitice autoimune, al tiroiditei. Aceasta nu exclude însă şi un răspuns autoimun T. Alte BAI au - clar - ca efector LyT, aşa cum este encefalomielita alergică.

100
După mecanismul imunopatogenetic BAI se clasifică astfel :
TIPUL II = Ac faţă de antigenele de suprafaţă sau matrice : Anemia hemolitică autoimună Purpură trombocitopenică autoimună Sindromul Goodpasture Pemphigus vulgaris RAA ("febra" acută hemoragică) Bolile determinate de anticorpi faţă de receptorii celulari (Boala Graves, Miastenia gravis, Diabetul insulino-rezistent, Hipoglicemia) TIPUL III = Boala cu complexe imune : Glomerulonefritele post-streptococice Poliarterita nodoasă LES TIPUL IV = Boală mediată de celulele T : DID Poliartrita reumatoidă (ARJ) Encefalomielita autoimună, Scleroza multiplă
 Bolile determinate de anticorpi faţă de receptorii celulari (Boala Graves, Miastenia gravis, Diabetul insulino-rezistent, Hipoglicemia) TIPUL III = Boala cu complexe imune : Glomerulonefritele post-streptococice. Poliarterita nodoasă. LES. TIPUL IV = Boală mediată de celulele T : DID. Poliartrita reumatoidă (ARJ) Encefalomielita autoimună, Scleroza multiplă.")
101
Teoriile moderne - bazate pe descoperiri recente - susţin că BAI sunt boli multifactoriale, care se datorează intervenţiei unor numeroşi factori : Imunologici Genetici Extrinseci (Foarte importantă intervenţia factorilor infecţioşi! ! !) Hormonală Psihologici Mecanismul care conduce la BAI (autoimunitate patologică) nu este univoc. Existenţa unui deficit imun (DI) favorizează apariţia manifestărilor AI, fapt susţinut de modelele animale şi umane.
 Hormonală. Psihologici. Mecanismul care conduce la BAI (autoimunitate patologică) nu este univoc. Existenţa unui deficit imun (DI) favorizează apariţia manifestărilor AI, fapt susţinut de modelele animale şi umane.")
102
A. Date experimentale Unele modele animale ilustrează clar rolul deficitului imun în apariţia manifestărilor autoimune. (A.M.Prieur, C.Griscelli) Astfel, şoarecele NZB dezvoltă spre vârsta de 6 luni o anemie hemolitică cu testul Coombs-pozitiv, prezenţa de AAN şi o glomerulonefrită cu depozite glomerulare de imunoglobuline şi AADNA. Un deficit timic precede această afecţiune, traducându-se prin atrofia epiteliului timic, pierderea funcţiilor LyTs - fenomene însoţite de o creştere a producţiei de Ac faţă de antigene timo-independente. În timp, se constată o diminuare tot mai accentuată a răspunsului la antigene solubile sau celulare şi dispariţia reacţiei grefonului împotriva gazdei. La vârsta de 12 luni, survine un limfom B terminal. Majoritatea anomaliilor descrise se corijează prin grefa timică efectuată precoce sau prin injectarede hormoni timici. Şoarecii Swan, prezentând o alterare a funcţiei timice, sunt caracterizaţi de o producţie crescută de AcAN şi de o glomerulonefrită cu complexe imune. Există numeroase modele animale, toate demonstrând posibilitatea evoluţiei de la un deficit imunitar la o hiperreactivitate intrinsecă a LyB, cu apariţia unor manifestări patologice de AI.

103
B. Date clinice O serie de modificări imunologice sugerează un posibil deficit imunologic în cursul afecţiunilor autoimune. Astfel, în cursul LED, unele anomalii constatate amintesc de cele din modelele animale. Producţia spontană de imunoglobuline in vitro de către LyB în cursul LED este cu mult mai crescută decât la indivizii normali. Aceasta ar putea fi consecinţa creşterii absolute sau relative a LyTh (CD4+) sau a diminuării funcţiei LyTs (CD8+). Prezenţa de Ac anti-limfocite supresoare susţine ipoteza anterioară, ca şi diminuarea producţiei de IL2 în LED. Este posibil ca pe fondul unui deficit minor al imunităţii să se dezvolte BAI la întâlnirea organismului cu unii factori antigenici extrinseci, de care organismul nu are mijloace de a se debarasa. În legătură cu aceasta, trebuie discutate două aspecte, şi anume fenomenul de creştere a nivelului Ac îndreptaţi împotriva diverselor microorganisme, şi rolul macrofagului.
 sau a diminuării funcţiei LyTs (CD8+). Prezenţa de Ac anti-limfocite supresoare susţine ipoteza anterioară, ca şi diminuarea producţiei de IL2 în LED. Este posibil ca pe fondul unui deficit minor al imunităţii să se dezvolte BAI la întâlnirea organismului cu unii factori antigenici extrinseci, de care organismul nu are mijloace de a se debarasa. În legătură cu aceasta, trebuie discutate două aspecte, şi anume fenomenul de creştere a nivelului Ac îndreptaţi împotriva diverselor microorganisme, şi rolul macrofagului.")
104
Creşterea nivelului anticorpilor îndreptaţi împotriva unor microorganisme este observată în cursul numeroaselor afecţiuni autoimune şi inflamatorii cronice, ceea ce ridică problema rolului posibil al unui agent infecţios la originea acestor afecţiuni şi a mecanismului prin care factorul infecţios ar persista în organism. Se cunoaşte faptul că o similitudine antigenică între peretele bacterian al Streptococului β-hemolitic şi ţesuturile cardiace reprezintă unul dintre factorii de apariţie ai RAA. De asemenea, o modificare structurală a celulelor HLA B 27+ de către o plasmidă provenind din Klebsiella, în cursul spondilitelor şi artritelor reacţionale este implicată în apariţia reumatismelor asociate cu HLA B27+. Printre virusurile incriminate în patogenia unor reumatisme inflamatorii cronice amintim virusul Rubeolei, Parvovirusul B19, virusul Epstein-Barr sau virusul Coxackie B în geneza dermatomiozitei.

105
S-a demonstrat că injectarea intraperitoneală de extract de Lactobacillus casei - un microorganism saprofit al florei intestinale la om - determină la mai multe linii de şoareci o boală inflamatorie cu leziuni vasculare asemănătoare cu cele din boala Kawasaki. Boala, însă, nu apare la şoarecele C3H/HeJ, caracterizat printr-un deficit funcţional al macrofagului. Aceste observaţii indică importanţa rolului macrofagului în apariţia leziunilor vasculare, ea putând ajuta şi la înţelegerea efectului spectacular al injecţiilor i.v. de imunoglobuline care - printre altele - acţionează prin saturarea receptorilor Fc ai macrofagelor la pacienţi.

106
MANIFESTĂRILE AUTOIMUNE ŞI INFLAMATORII OBSERVATE ÎN CURSUL DEFICITELOR CONGENITALE ALE SISTEMULUI IMUN Deficite imune înnăscute 1.1.Prezintă interes deficitele imunităţii umorale Pot exista hipogammaglobulinemii de expresie variabilă, cunoscute mai târziu decât agammaglobulinemia Bruton; sunt neereditare; grupează un ansamblu heterogen la care nivelul anomaliei de diferenţiere a liniei B este variabil. Rezultă un DI umoral profund, cu infecţii comparabile cu cele din boala Bruton.

107
Tratamentul cu Ig este uneori activ asupra acestor artrite.
În cursul DI umorale, pot apare manifestări articulare de tip ARJ la % dintre subiecţii cu hipogammaglobulinemie şi pot fi de tipul : poli/oligoartritelor, cu vârstă de debut variabilă. Se pot asocia noduli cutanaţi, iar bioptic se observă inflamaţie nespecifică, cu hiperplazie sinovială şi absenţa plasmocitelor. Nu există auto-Ac. Se impune diferenţierea faţă de artritele bacteriene, frecvente pe acest teren. Tratamentul cu Ig este uneori activ asupra acestor artrite. Patogenia acestor manifestări sugerează intervenţia diverselor mecanisme.

108
Pot să apară şi alte afecţiuni sistemice, amintind conectivitele.
Primul constă în prezenţa de microorganisme care persistă, pledând în favoarea acestui deficit, ceea ce s-a demonstrat pentru prima dată pentru Mycoplasma. Această etiologie are şi probe terapeutice, ajungându-se la ameliorare după tratament antibiotic adecvat. Au fost semnalaţi şi alţi agenţi infecţioşi, de exemplu virali. Pot să apară şi alte afecţiuni sistemice, amintind conectivitele. Tablouri clinice care sugerează o dermatomiozită sau o polimiozită, asociindu-se semne cutanate, edeme, slăbiciune musculară nu sunt excepţionale. Aceste manifestări pot fi asociate sau nu artritelor şi unei suferinţe meningoencefalitice non-bacteriene.

109
Uneori, apar manifestări digestive, cu atrofia mucoasei jejunale sau cu hipertrofie limfoidă (radiologic şi histologic). Ele pot apare în cursul hipogammaglobulinemiilor cu expresie variabilă. De asemenea, pot apare cointeresări hepatice cronice, cel mai adesea în contextul unei suferinţe pluriviscerale. Adeseori există manifestări hematologice : anemie hemolitică, lecopenii, trombocitopenii. Tulburările endocrine şi manifestările cu caracter alergic pot să apară, de asemenea, pe fondul DI.

110
Deficitele selective în Ig, la rândul lor, creează condiţii pentru instalarea bolilor autoimune.
Deficitul în IgA este cel mai frecvent, cu o incidenţă crescută a infecţiilor digestive şi în sfera ORL. S-au descris în deficitul în IgA şi manifestări autoimune : LED, ARJ, dermatomiozită, tiroidită, boala celiacă, boala Addison, enterita regională, colita ulceroasă, sindrom Sjögren, vascularite cerebrale, anemie pernicioasă, purpură trombocitopenică idiopatică. Deficitul selectiv în IgA poate fi primar sau dobândit. Poate fi în relaţie cu inducerea unei supresii T. În unele cazuri DI selectiv de IgA este tranzitoriu şi legat de o autoimunitate anti-IgA. Deficitul în IgA poate fi izolat, asociat deficitului de subclase de IgG sau IgE cu prezenţa unei IgM cu GM mică.

111
Pot să apară şi alte deficite selective :
Deficitul în subclase de Ig poate fi asociat cu alte deficite imune. Nu sunt excepţionale. Deficitul de IgG2 apare frecvent în patologia autoimună şi inflamatorie. Pot să apară şi alte deficite selective : În mod normal, raportul lanţurilor k/λ al Ig, este 2/1, el poate suferi modificări în sensul absenţei totale a lanţurilor k, apare la mai multe cazuri, evoluând cu fibroză chistică de pancreas. Disgammaglobulinemia de tip I (IgG şi IgA scăzute şi IgM crescut), situaţie în care sunt frecvente infecţiile bacteriene şi semnele ce evocă un proces inflamator cronic. Hipogammaglobulinemia cu timom : se observă absenţa sau scăderea marcată a celulelor B circulante. Sindromul se poate însoţi de manifestări autoimune. Timectomia este urmată de vindecare.
, situaţie în care sunt frecvente infecţiile bacteriene şi semnele ce evocă un proces inflamator cronic. Hipogammaglobulinemia cu timom : se observă absenţa sau scăderea marcată a celulelor B circulante. Sindromul se poate însoţi de manifestări autoimune. Timectomia este urmată de vindecare.")
112
1.2. Deficitele predominante pe funcţia T pot pregăti terenul pentru instalarea autoimunităţii.
Fenomene autoimune se observă în sindromul Wiskott-Aldrich (ereditar, recesiv, legat de cromozomul X, cu evoluţie spontană, în general mortală, asociind : eczemă, trombocitopenie, infecţii bacteriene recurente, cu apariţie precoce; DI vizează funcţia umorală, mai ales IgM, cu pierderea răspunsului la antigene polizaharidice şi DI celular progresiv). DI în ataxie - teleangiectazie (ereditară, autozomal-recesivă, asociind : ataxie cerebeloasă, teleangiectazie cutanată şi oculară şi - frecvent - infecţii ORL şi pulmonare în repetiţie) este de intensitate variabilă; este mai ales umoral, în special IgA şi progresiv celular; este unul din DI congenitale cele mai frecvente. În evoluţie apar tulburări neurologice majore, complicaţii legate de infecţii, riscul dezvoltării unor afecţiuni maligne (leucemii, limfom).
. DI în ataxie - teleangiectazie (ereditară, autozomal-recesivă, asociind : ataxie cerebeloasă, teleangiectazie cutanată şi oculară şi - frecvent - infecţii ORL şi pulmonare în repetiţie) este de intensitate variabilă; este mai ales umoral, în special IgA şi progresiv celular; este unul din DI congenitale cele mai frecvente. În evoluţie apar tulburări neurologice majore, complicaţii legate de infecţii, riscul dezvoltării unor afecţiuni maligne (leucemii, limfom).")
113
Manifestări autoimune în sdr
Manifestări autoimune în sdr. Wiskott-Aldrich şi în ataxie-teleangiectazie Fenomene AI se observă şi în candidoza cronică cutaneo - mucoasă (DI predominant celular). Candidoza este gravă; cuprinde treptat învelişul cutaneo - mucos; la un caz din două survine o endocrinopatie de natură AI (Boală Addison, hipoparatiroidism, hipotiroidism, hipogonadism, diabet) + DI selectiv în răspunsul celular faţă de Candida.
. Candidoza este gravă; cuprinde treptat învelişul cutaneo - mucos; la un caz din două survine o endocrinopatie de natură AI (Boală Addison, hipoparatiroidism, hipotiroidism, hipogonadism, diabet) + DI selectiv în răspunsul celular faţă de Candida.")
114
1.3. Tulburări autoimune pot să survină şi în DI ale compartimentului nespecific. Anomaliile polinuclearelor sunt numeroase : determină infecţii şi unele pot fi asociate -direct sau nu - cu fenomene AI. În granulomatoza sistemică cronică, există o anomalie intrinsecă a polinuclearelor, cu scăderea bactericidiei faţă de diverşi germeni : Stafilococ, Serratia, Aspergillus. Substratul este o tulburare a metabolismului oxidativ. Infecţiile se localizează la nivelul ganglionilor, ficatului, plămânilor, cu formarea de abcese; are un tratament dificil. Este o anomalie recesivă, legată de cromozomul X. La 30 % din mame se observă manifestări lupice cutanate, cu prezenţa AAN. Deficitul incriminat este insuficient pentru a determina o infecţie, dar nu permite catabolismul complet al antigenelor; creşte retenţia antigenică urmată de autoimunizare. În neutropeniile constituţionale apariţia semnelor digestive neinfecţioase trebuie să conducă la căutarea unei posibile boli Crohn.

115
1.4. Deficitele sistemului complement pot "determina" şi infecţii, dar ele sunt asociate cu manifestări AI. În deficitele căii clasice de activare a complementului, la 2/3 din cazuri au fost observate conectivite. LED şi vascularitele cronice sunt asociate mai ales deficitului homozigot în C4. Deficitele heterozigote de C4 din cursul LED ar interesa 3/4 din părinţi. Deficitul homozigot în C2 este frecvent la subiecţi cu infecţii repetate şi BAI. Manifestările clinice observate în deficitul homozigot în C2 sunt : LED, glomerulonefrita, purpura reumatoidă, dermatomiozita, vascularita cronică, boala Crohn, tulburări de agregare plachetară, boala Hodgkin, DI variabil, infecţii repetate.

116
Deficite imune dobândite
În cursul DI dobândite (mai ales în cursul infecţiilor virale), manifestările AI pot fi pe primul plan. Multe virusuri conduc la DI tranzitorii, în cursul cărora creşte riscul dezvoltării infecţiilor. Este cunoscută puterea anergizantă a virusului rujeolic. Alte virusuri ce pot determina DI sunt : v.gripal, v.rubeolei, v.citomegalic, v.Epstein-Barr. Sunt semnalate uneori manifestări "reacţionale" tranzitorii (artrite, semne cutanate). Ele pot dura săptămâni, pot regresa spontan. Mecanismul este - încă - neelucidat. Ar fi implicate acţiunea complexelor imune, analogia structurală între antigenele virusului şi ale gazdei sau perturbarea reţelei citokinelor. În infecţia cu HIV, poate să apară un sindrom ce evocă LED şi care poate constitui prima manifestare a bolii cu evidenţierea a numeroşi auto-anticorpi : anti-eritrocitari, AAN, anti-trombocitari şi scăderea complementului.
, manifestările AI pot fi pe primul plan. Multe virusuri conduc la DI tranzitorii, în cursul cărora creşte riscul dezvoltării infecţiilor. Este cunoscută puterea anergizantă a virusului rujeolic. Alte virusuri ce pot determina DI sunt : v.gripal, v.rubeolei, v.citomegalic, v.Epstein-Barr. Sunt semnalate uneori manifestări reacţionale tranzitorii (artrite, semne cutanate). Ele pot dura săptămâni, pot regresa spontan. Mecanismul este - încă - neelucidat. Ar fi implicate acţiunea complexelor imune, analogia structurală între antigenele virusului şi ale gazdei sau perturbarea reţelei citokinelor. În infecţia cu HIV, poate să apară un sindrom ce evocă LED şi care poate constitui prima manifestare a bolii cu evidenţierea a numeroşi auto-anticorpi : anti-eritrocitari, AAN, anti-trombocitari şi scăderea complementului.")
117
Problema tratamentului bolilor autoimune este deosebit de dificilă
Problema tratamentului bolilor autoimune este deosebit de dificilă. Posibilităţile actuale şi de perspectivă ale tratamentului sunt sintetizate în următoarea schemă : (după Roitt, Brostoff, Male) În ceea ce priveşte DI, ele : - trebuie depistate şi urmărite în evoluţie - trebuie tratate prin utilizarea imunomodulatorilor stimulanţi, cunoscându-se azi mult mai bine direcţiile de acţiune ale acestora, favorizând astfel ameliorarea sau normalizarea reacţiilor imune, cu răspuns mai eficient faţă de agresorii infecţioşi şi împiedicând declanşarea unei AI manifeste pe fondul deficitului imun.
 În ceea ce priveşte DI, ele : - trebuie depistate şi urmărite în evoluţie. - trebuie tratate prin utilizarea imunomodulatorilor stimulanţi, cunoscându-se azi mult mai bine direcţiile de acţiune ale acestora, favorizând astfel ameliorarea sau normalizarea reacţiilor imune, cu răspuns mai eficient faţă de agresorii infecţioşi şi împiedicând declanşarea unei AI manifeste pe fondul deficitului imun.")
118
DIRECŢIILE DE ACŢIUNE A DIVERŞILOR IMUNOMODULATORI

Παρόμοιες παρουσιάσεις

















>")






