Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
DIFERENTIEREA SI MATURAREA SEXUALA: BAZE GENETICE, ENDOCRINOLOGIE, ETAPE

2
Sexualizarea normală presupune diferenţierea unor structuri şi funcţiuni care fac organismul apt de procreere. Diferenţierea structurilor sexuale este un adevărat act de morfogeneză, deoarece din elemente ale organismului nediferenţiate şi iniţial ambigene iau naştere structuri unisexuate, masculine sau feminine. Această diferenţiere este determinată de anumiţi factori sexualizanţi -, care prelucrează şi direcţionează într-un sens determinant şi unigen sexualizarea organismului. (C. Dumitrache)
")
3
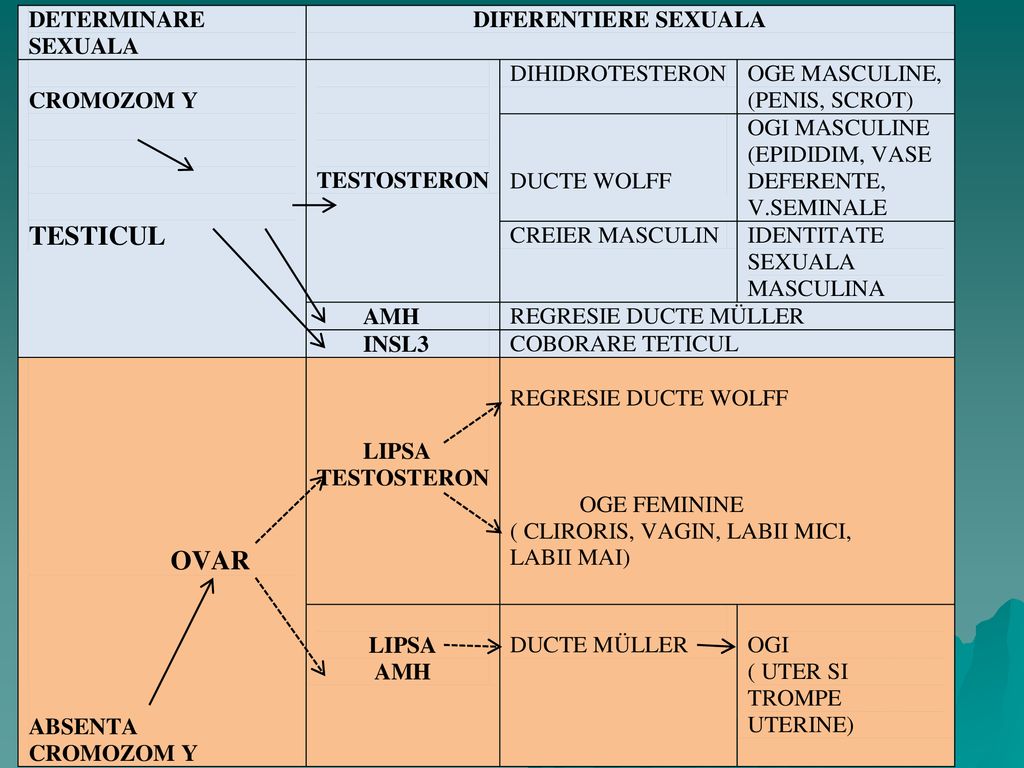
DETERMINAREA SEXUALA NORMALA ŞI DIFERENŢIEREA SEXUALA Determinarea şi diferenţierea sexului reprezintă procese secvenţiale care implica: - asezari succesive ale cromozomului sexual în zigot în momentul concepţiei, - determinarea sexului gonadal (primar) prin sex genetic - organizarea sexului gonadal pentru diferenţierea aparatului genital şi de aici determinarea sexului fenotipic. La pubertate, dezvoltarea caracterelor sexuale secundare specifice reintareste şi face să devină mult mai vizibile manifestările fenotipice ale dismorfismului sexual.

4
Ontogenia caracteristicilor sexuale
Identificare Origine Factori determinanti Sex cromozomial Analiza cariotipului Cromozomii sexuali parentali Normal: compozitia cromozomiala a spermei Anormal: Nondisjuncţii ale diviziunii meiotice Cromatina X Neutrofile frotiu / sectiuni din alte tesuturi Heterocroma- tinizarea cromozom X Inactivare parţiala şi formarea heterocromatinei în toţi cromozomii X Corpusculul Y Idem Cromozom Y Segmentul distal heterocromatic – braţ lung Y

5
Sex gonadal Aspect clinic Testicul Ovar Testicul: gena SRY şi gene autozomale Ovar: gene de pe cromozomul X; gene germinale şi autozomale Ducte genitale Ex.clinic şi imagistic pelvic Ducte mulleriene şi wolffiene Tendinta intrinseca la feminizare Organe genitale externe Ex. clinic, uretroscopie – investig. sinus urogenital şi/sau Rx Tuberculi genitali, pliu uretral, la bioscrot, sinus urogenital Tendinta intrinseca la feminizare; masculinizarea necesita stimulare androgenica inainte de sapt. a 12-a fetală

6
Sex hormonal Caractere sex.secundare: masculine: pilozitate, voce, musculatură, dimensiuni falus feminine: sâni, menstruatie, ovulatie, tract reproductiv Pattern hormonal -masculin: testosteron, FSH/LH -feminin: secreţie ciclica FSH / LH, estrogeni, progesteron Hipotalamus: LHRH Hipofiză Celule secretorii din testicul, ovar, suprarenală Hipotalamus: LHRH Hipofiză: LH/FSH – secreţie pulsatilă de LHRH, steroizi gonadali şi inhibină Gonade: diferenţierea cel. Secretorii şi biosinteza enzimatică; stimularea prin FSH/LH Sensibilitate de organ

7
Identitatea de gen Identitate ca bărbat sau femeie Apariţia OGE, fact. de mediu, androgeni prenatali, gene, catactere sexuale pubertare Expunere prenatală la androgenii din testiculi – contribuie la identitatea de sex masculin Factori psihologici în primii ani: atitudinea părinţilor, interacţiunea cu ambele sexe, caractere sexuale secundare la pubertate, gene – x-linkat şi y-linkat

8
Morfogeneza structurilor sexuale Sexualizarea organismului este un proces care se desfăşoară progresiv, după un program prestabilit genetic. Ordinea de desfăşurare a principalelor evenimente sexuale este următoarea: - formarea structurii sexuale cromozomiale sau sexualizarea genetică (Gz); - formarea organului producător de gameti şi de hormoni sexoizi, gonada (Go); - formarea canalelor de evacuare a gametilor, ductele genitale interne sau organele genitale interne (OGI);
; - formarea organului producător de gameti şi de hormoni sexoizi, gonada (Go); - formarea canalelor de evacuare a gametilor, ductele genitale interne sau organele genitale interne (OGI); .")
9
formarea organelor genitale externe;
stabilirea sexualizării nervoase şi comportamentale, sexualizarea neuro-comportamentală (NC); sexualizarea puberală (Pub.); “În timpul acestor “etape” ale sexualizării au loc fenomene sexualizante complexe”.
; sexualizarea puberală (Pub.); În timpul acestor etape ale sexualizării au loc fenomene sexualizante complexe .")
10
Etapele sexualizării Structualizarea sexului genetic
Structualizarea sexului este consecinta formulei cromozomiale a gametilor participanti la actul fecundaţiei Sexul genetic – este nu numai prima diferenţiere sexuală a individului dar şi prima diferenţiere fenotipică Structuralizarea gonadei Formarea gonadei unisexuate este progresivă, având ca punct de plecare un organ cu particularităţi structurale care îi permit să devină în final tot atât de bine testicul sau ovar.

11
Etapa gonadogenezei 1. Formarea mugurelui germinativ sau a crestei germinale 2. Formarea gonadei nediferenţiate sau, mai precis, a gonadei bisexuate 3. Formarea gonadei unisexuată (ca ovar sau testicul, este rezultatul diferenţierii sexuale diferite a unora şi aceloraşi celule, iniţial sexualixeşte bipotenţiale). Desi în gonadele de sex diferit sunt diferenţiate morfologic specific, aceste celule au caractere funcţionale omoloage; spermatogoniile şi ovogoniile asigură gametogeneza, celulele Sertoli şi granuloase sun elemente trofice şi secretorii, celulele interstiţiale sun hormonogenetice.
. Desi în gonadele de sex diferit sunt diferenţiate morfologic specific, aceste celule au caractere funcţionale omoloage; spermatogoniile şi ovogoniile asigură gametogeneza, celulele Sertoli şi granuloase sun elemente trofice şi secretorii, celulele interstiţiale sun hormonogenetice.")
12
Structualizarea organelor genitale interne
Este reprezentată de formarea organelor genitale interne de tip masculin sau feminin Diferenţierea unisexuată porneşte dela structuri ambisexuale, ceea ce permite diviziunea etapei in doua subetape distincte: a structurilor ambigene a organelor genitale interne unisexuate

13
SEXUL CROMOZOMIAL SI CROMOZOMII X ŞI Y

14
CROMOZOMUL Y

15
STRUCTURA CROMOZOMULUI Y
2% din genomul uman şi are 60Mb reg.eucromatică -genetic activă -are 2 zone- segment Y-specific - regiunile pseudoautozomale PAR1(cu 10 gene)/PAR 2 (cu 4 gene)- braţul scurt terminal şi braţul lung, omologe crom. X reg.heterocromatică- genetic inactivă - aparţine braţului lung
/PAR 2 (cu 4 gene)- braţul scurt terminal şi braţul lung, omologe crom. X. reg.heterocromatică- genetic inactivă. - aparţine braţului lung.")
16
Funcţiile biologice ale cromozomului Y:
-dezv. testiculară-gene masculin-determinante, indiferent de nr. cr. X -spermatogeneză

17
Mecanismul anomaliilor cromozomiale
Aneuploidismul – celulele aneuploide conţin un număr de cromozomi diferit din punct de vedere al caracteristicilor şi al speciei. Mozaicismul – mozaicismul este prezent individual sau .... Chimerismul – există individual sau de doua ori mai multe celule liniare

18
Erorile structurale Deletia Duplicarea Translocarea
Sunetul cromozomilor

19
Cromatina Y: -fluorocrom quinanacrin hidroclorura-identifică cromatina Y în metafază -FISH-identifică cromoz. Y în interfază

20
Genele şi organogeneza testiculelor

21
Antigenul H-Y iniţial-considerat un factor de determinare testiculară lipsa reproductibilităţii gena Ag H-Y- pe braţul lung al crom. Y, distinct de gena determinării sexuale masculine -Gena ZFY localizare- braţul scurt al crom. Y 1987 Page şi colab.- posibil rol determinarea sexuală, infirmat ulterior( prezenţa şi pe crom.X, care scapă inactivării)
")
22
- Gena SRY: identificată în 1989 de Palmer
localizare-braţul scurt al crom. Y este factorul determinant al dezv. testiculare- studii efectuate pe şoareci subiecţii de sex masculin, SRY-inactivi- ambiguitate a OGE şi hermafroditism adevărat cele mai frecv. mutaţii-reg. HMG 4 roluri-inducerea diferenţierii cel. Sertoli -migrarea cel. mezonefrice în creasta genitală -proliferarea cel. creastei genitale -vascularizaţie -recrutare de cel. endoteliale din mezonefros -

23
LOCALIZAREA SRY LA NIVELUL CROMOZOMULUI Y(BRAŢ SCURT)
")
24
ALTE GENE IMPLICATE ÎN DETERMINAREA TESTICULARE

26
GENE CU ROL ÎN DEZV. TESTICULARĂ

27
Gena WT1 (gena supresoare a tumorii Wilms)
codifică un factor de transcripţie Localizare: cr. 11p13 Rol important în determinarea şi diferenţierea sexuala – studii pe şoareci şi oameni Mutaţii heterozigote în exoni sdr. Denys-Drash (pseudohermafroditism masculin + tumoră Wilms) Mutaţii heterozigote + pierderea izoformei KTS sdr. Frasier (disgenezie gonadală XY) Deleţie sdr. WAGR (ambiguitate genitală)
 Mutaţii heterozigote + pierderea izoformei KTS sdr. Frasier (disgenezie gonadală XY) Deleţie sdr. WAGR (ambiguitate genitală)")
28
Gena SF1 (factorul steroidogenic 1)
codifică receptorul nuclear orfan şi un factor de transcripţie Localizare: - cr. 9q33 Mutaţii heterozigote la sexul XY- anularea sexualizării + insuficienţă adrenală la sex XX – insuficienţă adrenală + ovare normale Mutaţii homozigote anularea sexualităţii XY + insuficienţă adrenală

29
Gena SOX9 Codifică factorul de transcripţie Localizare: cr.17q24
Afectarea ei pseudohermafroditism XY Duplicarea ei mascul XX

30
Gena DMRT1 Codifică factori de transcripţie Localizare: cr. 9p24,3
Deleţia 9p24,3 pseudohermafroditism masculin XY, retard mental. femeile XX au funcţie ovariană variabilă

31
Gena DAX1 codifică receptorul nuclear orfan şi un factor de transcripţie Localizare: cr. Xp21,3 Mutaţii XY (diferenţierea testiculară normală), hipoplazie adrenală, hipogonadism hipogonadotrop Duplicarea pseudohermafroditism masculin XY
, hipoplazie adrenală, hipogonadism hipogonadotrop. Duplicarea pseudohermafroditism masculin XY.")
32
Gena WNT4 Gena ATRX Codifică molecule de semnal Localizare: cr. 1p35
Duplicarea pseudohermafroditism masculin XY Gena ATRX Codifică enzime = helicaze Localizare : cr. Xq13,3 Deleţii şi mutaţii pseudohermafroditism masculin, talasemia, retard mental

33
Cromozomul 10q- Gena FOXL2
Deleţii terminale anomalii urogenitale: hipoplazie renală, criptorhidism, micropenis, hipospadias şi, rareori, anularea sexualizării XY Gena FOXL2 Codifică factori de transcripţie Localizare: cr. 3q23 Mutaţii blefarofimoză/ epicantus / ptoză/fertilitatea masculină se menţine

34
DHH Codifică molecule semnal Localizare: cr. 12q13.1
Mutaţii homozigote disgenezie gonadală parţială + neuropatie minifasciculară

35
CROMOZOMUL X-STRUCTURĂ

36
CROMOZOMUL X-STRUCTURĂ
Braţ scurt-PAR1(reg. pseudoautozomală), omoloagă cr. Y -gene cu rol în determinarea şi diferenţierea sexuala:gena sindr Kallmann, DMD, DAX1 Braţ lung-gene inactivatoare Reg. centromerică-gena receptorului androgenic
, omoloagă cr. Y. -gene cu rol în determinarea şi diferenţierea sexuala:gena sindr. Kallmann, DMD, DAX1. Braţ lung-gene inactivatoare. Reg. centromerică-gena receptorului androgenic.")
37
Funcţiile biologice ale cromozomului X
-mai complexe decât ale crom. Y(are 160Mb): determinarea sexuală atât laambele sexe Diferenţierea structurilor somatice la bărbat Diferenţierea normală ovariană şi maturarea foliculară - necesită prezenţa ambilor cr. X
: determinarea sexuală atât laambele sexe. Diferenţierea structurilor somatice la bărbat. Diferenţierea normală ovariană şi maturarea foliculară - necesită prezenţa ambilor cr. X.")
38
Cromatina X(corpuscul Barr):
-descris de Barr şi Bertram în 1949-masă de cromatină la periferia nucleului -se determină din frotiu realizat din mucoasa bucală. În anumite ţesuturi aproape toţi nucleii sânt cromatino-pozitivi( ex. lichidul amniotic) -nr. de corpusculi Barr este cu unul mai puţin decât nr. total de crom.X -anomaliile formei şi dimensiunii cromatinei X se corelează cu anomalile crom.X
 -nr. de corpusculi Barr este cu unul mai puţin decât nr. total de crom.X. -anomaliile formei şi dimensiunii cromatinei X se corelează cu anomalile crom.X.")
39
CROMATINA X PE FROTIU DE MUCOASĂ BUCALĂ LA O FEMEIE NORMALĂ

40
CROMATINA X ŞI EXPRESIA GENELOR
doar unul din cei 2 crom. X este activ în interfază inactivarea crom. X- faza tardivă de blastocit( între zilele 12-18) inactivarea –mai multe etape: 1. identificarea crom. X 2. selectarea aleatorie a crom. X materni sau paterni pt. Inactivare 3. menţinerea inactivării
 inactivarea –mai multe etape: 1. identificarea crom. X. 2. selectarea aleatorie a crom. X materni sau paterni pt. Inactivare. 3. menţinerea inactivării.")
41
GENELE DIN ORGANOGENEZA OVARIANĂ
Gene din familia WNT: -Wnt4-este,,down-regulation,,în testicul -prezentă în mezenchimul ce formează ductele mulleriene -supresia diferenţierii cel. Leydig în ovar -iniţiază dezv. ductului mullerian ambele sexe -Wnt7-complează diferenţierea ductelor mulleriene–>uter, trompe, 1/3 sup. vagin

42
DIFERENTIEREA OGI ŞI OGE

43
DIFERENTIEREA OGE FEMININE SI MASCULINE DIN GONADA PRIMORDIALA
44
DIFERENTIEREA PSIHOSOCIALA
Identitate sexuala ( masculin sau feminin) Rol sexual ( comportament) Orientare sexuala ( alegerea partenerului) Abilitati cognitive specifice
 Rol sexual ( comportament) Orientare sexuala ( alegerea partenerului) Abilitati cognitive specifice.")
46
INTERSEXUALITATEA TULBURARI ALE DEZVOLTARII SEXUALE(DSD) Termenul de Disorders of sex development (DSDs) il va inlocui pe acela de pseudohermafrodism ( in urma consensului de la Chicago, 2006) termen, care desemna o sexualizare deficitara masculina- subvirilizare in cazul pseudohermafroditismului masculin ( la subiecti cu cariotip 46 XY) , masculinizare, virilizare in cazul pseudohermafroditismului feminin ( la femei, cu cariotip 46XX) sau ovotestis – hermafroditism adevarat
 Termenul de Disorders of sex development (DSDs) il va inlocui pe acela de pseudohermafrodism ( in urma consensului de la Chicago, 2006) termen, care desemna o sexualizare deficitara masculina- subvirilizare in cazul pseudohermafroditismului masculin ( la subiecti cu cariotip 46 XY) , masculinizare, virilizare in cazul pseudohermafroditismului feminin ( la femei, cu cariotip 46XX) sau ovotestis – hermafroditism adevarat")
47
CLASIFICAREA PSEUDOHERMAFRODITISMULUI FEMININ
I. -Indus de Androgeni Sursa Fetala 1. Hiperplazie adrenala Congenitala a. Virilism numai, 21-hidroxilare adrenala defectiva (CYP21) b. Virilism sindrom cu pierdere de sare, deficit de 21 hidroxilaza adrenala (CYP21) c. Virilism cu hipertensiune arteriala, deficit 11β hidroxilaza(CYP11B1) d. Virilism cu insuficienta adrenala,deficit de 3-HSD 2 (HSD3B 2) 2. Deficit de P450 aromataza (CYP19) 3. Mutatia genei receptorului de Glucocorticoizi B. Sursa Materna 1. Iatrogen a. Testosteron si steroizi inruditi b. Progestageni sintetici orali, rar diethylstilbestrol 2. Tumori Virilizante ovariene sau adrenale 3. Luteomul virilizant de sarcina 4. Hiperplazie adrenala congenitala, virilizanta a mamei C. Sursa nedeterminata 1. ? . Luteomul virilizant de sarcina II. Modificari Non–Androgen-Induse a Diferentierii Structurilor Urogenitale
 b. Virilism sindrom cu pierdere de sare, deficit de 21 hidroxilaza adrenala (CYP21) c. Virilism cu hipertensiune arteriala, deficit 11β hidroxilaza(CYP11B1) d. Virilism cu insuficienta adrenala,deficit de 3-HSD 2 (HSD3B 2) 2. Deficit de P450 aromataza (CYP19) 3. Mutatia genei receptorului de Glucocorticoizi. B. Sursa Materna. 1. Iatrogen. a. Testosteron si steroizi inruditi. b. Progestageni sintetici orali, rar diethylstilbestrol. 2. Tumori Virilizante ovariene sau adrenale. 3. Luteomul virilizant de sarcina. 4. Hiperplazie adrenala congenitala, virilizanta a mamei. C. Sursa nedeterminata Luteomul virilizant de sarcina. II. Modificari Non–Androgen-Induse a Diferentierii Structurilor Urogenitale.")
48
CLASIFICAREA PSEUDOHERMAFRODITISMULUI MASCULIN
I. Pseudohermafroditism masculin A. Testicul rezistent la hCG and LH (agenezia celulelor Leydig sau hipoplazia determinata de defect al receptorului hCG/LH) B. Erori inascute alebiosintezei de testosteron 1. Deficite enzimatice care afecteaza sinteza atat a corticosteroizilor cat si a testosteronului (variante de hiperplazie adrenala congenitala) a. Deficit de StAR (hiperplazie adrenala congenitala lipoida) b. Deficit de clivare a lantului lateral (P450scc), heterozigot c. Deficit de3-Hidroxisteroid dehidrogenaza/4,5 -izomeraza tipul II (3-HSD II) d. Deficit CYP17 (P450c17 [17-hydroxylase/17,20-lyase]) e. Deficit de 7-dehidrocolesterol reductaza Smith-Lemli-Opitz sindrom 2. Defecte enzimatice care afecteaza biosinteza de testosteron de catre testicule a. Deficit de CYP17 (P450c17 [17,20 liaza]) b. Deficit de 17-Hidroxisteroid dehidrogenaza tipe 3 (17-HSD 3)
 B. Erori inascute alebiosintezei de testosteron. 1. Deficite enzimatice care afecteaza sinteza atat a corticosteroizilor cat si a testosteronului (variante de hiperplazie adrenala congenitala) a. Deficit de StAR (hiperplazie adrenala congenitala lipoida) b. Deficit de clivare a lantului lateral (P450scc), heterozigot. c. Deficit de3-Hidroxisteroid dehidrogenaza/4,5 -izomeraza tipul. II (3-HSD II) d. Deficit CYP17 (P450c17 [17-hydroxylase/17,20-lyase]) e. Deficit de 7-dehidrocolesterol reductaza Smith-Lemli-Opitz sindrom. 2. Defecte enzimatice care afecteaza biosinteza de testosteron de catre testicule. a. Deficit de CYP17 (P450c17 [17,20 liaza]) b. Deficit de 17-Hidroxisteroid dehidrogenaza tipe 3 (17-HSD 3)")
49
C. Defecte in tesuturile tinta dependente de androgeni
1. Rezistenta la hormonii androgeni a. Sindromul de rezistenta completa la androgeni si variantele sale (testicul feminizant si variante) b. Sindromul de rezistenta incompleta la androgeni si variantele sale (sindrom Reifenstein) c. rezistenta la androgeni la barbati normali fenotipic (infertili si fertili) 2. Defecte in metabolismul testosteronului de catre tesuturile periferice ; Deficit de 5-reductaza-2 (SRD5A2) (pseudovaginal perineoscrotal hipospadias) D. Pseudohermafroditismul masculin Disgenetic 1. Disgenezia gonadica XY (incompleta) 2. XO/XY mozaicism,cromozom Y anormal structural, Mutatie SRY 3. Sindrom Denys-Drash (mutatie WT1) 4. Sindrom Frasier (mutatie a WT1 la jonctiune, mutatii-deletii KTS) 5. Sindrom WAGR ( deletie WT1 ) 6. Displazia Campomelica (mutatie SOX9) 7. Mutatie SFI
 b. Sindromul de rezistenta incompleta la androgeni si variantele sale (sindrom Reifenstein) c. rezistenta la androgeni la barbati normali fenotipic (infertili. si fertili) 2. Defecte in metabolismul testosteronului de catre tesuturile periferice ; Deficit de 5-reductaza-2 (SRD5A2) (pseudovaginal perineoscrotal. hipospadias) D. Pseudohermafroditismul masculin Disgenetic. 1. Disgenezia gonadica XY (incompleta) 2. XO/XY mozaicism,cromozom Y anormal structural, Mutatie SRY. 3. Sindrom Denys-Drash (mutatie WT1) 4. Sindrom Frasier (mutatie a WT1 la jonctiune, mutatii-deletii KTS) 5. Sindrom WAGR ( deletie WT1 ) 6. Displazia Campomelica (mutatie SOX9) 7. Mutatie SFI.")
50
8. DAX1 (duplicatie) 9. WNT4 (duplicatie) 10. 9p (DMRT1 deletie) 11. 10q 12. Sindrom ATRX (XH2 mutatie) 13. Sindromul de regresie testiculara E. Defecte in sinteza,secretia, sau raspunsul la Hormonul antimullerian: sindromul de duct mullerian persistent (Ducte genitale feminine la barbati normali; herniae uteri inguinale) F. Ingestie Materna de progestageni G. Produse chimice din mediu (endocrine disrupters) II. Forme neclasificate de dezvoltare sexuala anormala A. La barbati 1. Hipospadias 2. Organe genitale ambigue la barbati XY cu anomalii congenitale multiple B. La femei, absenta sau anomalii de dezvoltare a vaginului, uterului,si a trompelor uterine (sindromRokitansky-Kunster)
 F. Ingestie Materna de progestageni. G. Produse chimice din mediu (endocrine disrupters) II. Forme neclasificate de dezvoltare sexuala anormala. A. La barbati. 1. Hipospadias. 2. Organe genitale ambigue la barbati XY cu anomalii congenitale multiple. B. La femei, absenta sau anomalii de dezvoltare a vaginului, uterului,si a trompelor uterine (sindromRokitansky-Kunster)")
51
Insuficienţa gonadică determină o simptomatologie clinică ce depinde de:
tipul gonadei vârsta la care survine insuficienţa gonadică determinismul insuficienţei gonadice

52
Manifestări pubertare şi postpubertare în insuficienţa gonadică
Semnele insuficienţei gonadale pubertare şi postpubertare depind de sexul gonadei afectate (testicul sau ovar) şi de intensitatea insuficienţei (agonadism sau hipogonadism).
 şi de intensitatea insuficienţei (agonadism sau hipogonadism).")
53
FORME CLINICE CU FENOTIP MASCULIN SINDROMUL KLINEFELTER

54
Este o disgenezie orhitică ce devine manifestă clinic la pubertate
Vârsta pubertăţii este atinsă şi depăşită, fără ca fenomenele de sexualizare pubertară să apară sau, dacă au loc manifestări de masculinizare, ele sunt tardive şi incomplete Cauza sindromului este reprezentata de non-disjuncţia cromozomială a perechii de cromozomi X.

55
Frecvenţa sindromului Klinefelter este de
1:500-1:1000 de nou-născuţi de sex masculin şi se asociază cu vârsta crescută a mamei la naştere Morfotipul sexual îmbracă una din formele caracteristice insuficienţei orhitice: eunucoid, macroskel, hipoandric sau ginoid Cele mai frecvente sunt forma eunucoidă şi cea hipoandrica

56
In formele ginoide sau macroskele, paniculul adipos este relativ bine dezvoltat, cu o distribuţie feminoidă Musculatura este slab dezvoltată, vocea neformată, tegumentele palide, subţiri, delicate

57
Simptome Ginecomastia – frecventă dar nu obligatorie
Apare la pubertate sau după aceea, de obicei , bilateral, simetric sau asimetric; poate fi şi unilaterală Volumul variază de la acela al unui placard lenticular submamelonar la cel al unei portocale

58
Organele genitale externe sunt masculine, penisul infantil, subdezvoltat sau normal
Scrotul este uneori flasc, alteori redus ca volum, cu reflexul cremasterian diminuat sau abolit Testiculele sunt foarte mici – sub 1 cm -, de obicei dure, uneori moi şi întotdeauna insensibile la presiune; în contrast cu dimensiunile reduse, epididimul este aproximativ normal dezvoltat

59
Dezvoltarea psiho-intelectuală deficitară face ca procesul de şcolarizare să decurgă greoi; indivizii sunt însă apţi să înveţe o meserie Pulsiunile sexuale, deşi absente sau mult diminuate, nu sunt recunoscute, din contră, exagerate verbal

60
Clasificare noua a simptomatologiei
Simptomatologie fizica Simptomatologie sociala si de comportament simptomatologie de limbaj si a capacitatii de invatare Simptomatologie poli-x KS

61
Semiologia de laborator
Evidenţiază insuficienţa gonadică primitivă Valorile testosteronului plasmatic şi urinar sunt mult scăzute faţă de normal Gonadotrofinele hipofizare urinare sunt crescute Cromatina sexuală (testul Barr) este pozitivă; excepţional negativă, situaţie în care trebuie suspectat un mozaicism cromozomial Cariotip: 47XXY
 este pozitivă; excepţional negativă, situaţie în care trebuie suspectat un mozaicism cromozomial. Cariotip: 47XXY.")
62
Examenul bioptic al ţesutului gonadal arată ca leziuni esenţiale: hialinizarea tubilor seminiferi cu alterarea liniilor de spermatogeneză, fibrozarea ţesutului intertubular cu celule Leyding puţine, dispersate sau aglomerate în microadenoame Azoospermie

63
INSUFICIENŢELE OVARIENE

64
SINDROMUL TURNER FEMININ

65
Este un sindrom pluriformativ cu insuficienţă ovariană severă, în forma sa clasică, dar cu numeroase variante atât ale sindromului malformatic, cât şi ale insuficienţei ovariene Frecvenţa sindroMului este de 1 : nou născuţi de sex feminin

66
Forma “clasică” a sindromului Turner
Se defineşte prin patru elemente fundamentale: Hipotrofie staturală marcată şi disarmonică Malformaţii somatice numeroase şi grave Insuficienţă ovariană severă prin anovarie Cariotipul modificat specific şa frecvenţă: 45, XO

67
Morfotipul este caracterizat de:
Hipotrofia staturală marcată, cu valori medii ale înălţimii variind în jurul a 140 cm Aspect disarmonic şi îndesat dat de gâtul scurt, toracele lat, trunchiul ceva mai alungit comparativ cu membrele inferioare, scurte

68
Sindromul malformativ se prezintă astfel:
- Extremitatea cefalică: Frunte îngustă şi lată Inserţia joasă a părului pe frunte şi ceafă Urechi jos situate, uneori cu lobul urechii sudat Ochi oblicei, cu aspect mongoloid sau antimongoloid, putând prezenta epicantus, ptoză palpebrală, mistagmus, sclere albastre Nas înfundat la rădăcină Anomalii buco-dentare: comisurile labiale traseîn jos, palatul înalt boltit în ogivă, malocluzie dentară, dinţi supranumerari sau lipsă

69
Membre: Cubitus valgus: deviaţia antebraţului în afara axei braţului în poziţia de extensie şi supinaţie a membrului superior Degetele IV şi V scurtate la mâini şi la picioare Sindactilie, uneori

70
Gâtul, scurt şi gros, prezintă:
Pterygium colli: fald musculo-tegumentar întins în evantai pe feţele latero-posterioare ale gâtului, de la apofizele mastoide la umeri – “gât de sfinx” Toracele larg şi malformat de tipul “în carenă”, în scut”, “în butoi”; mameloane îndepărtate

71
Viscerele: Cordul : comunicaţii interatriale, interventriculare, coarctaţia aortei, stenoza istmului aortic, hipertensiune arterială Rinichiul: rinichi unic, rinichi în potcoavă, rinichi chistic, rinichi supranumerar, ureter dublu etc.

72
Tulburări senzoriale:
Anomalii cutanate: Nevi pigmentari răspândiţi pe faţă sau corp Tulburări senzoriale: Hipoacuzie Retinită pigmentară sau cataractă (mai rar)
")
73
Examenul radiologic evidenţiază o serie de alte malformaţii scheletice, cum ar fi:
Persistenţa cartilajelor de creştere după vârsta de închidere Brahimetacarpia şi brahimetatarsia IV şi V (semnul Archibald) Sinostoza radio-cubitală Distrofii vertebrale: cifoscolioza, blocuri vertebrale Osteoporoza generalizată sau localizată la vertebre, bazin etc.
 Sinostoza radio-cubitală. Distrofii vertebrale: cifoscolioza, blocuri vertebrale. Osteoporoza generalizată sau localizată la vertebre, bazin etc.")
74
Sindromul ovarian constă din:
Sexualizarea pubertară absentă cu: Absenţa sau foarte slaba dezvoltare a pilozităţii pubo-axilare Amastie Amenoree primară Organe genitale externe feminine sau infantile

75
Laparotomia exploratorie evidenţiază:
Dozările hormonale: Valorile sanguine şi urinare ale hormonilor ovarieni sunt, practic, nule; gonadotrofinele sunt crescute Laparotomia exploratorie evidenţiază: Organe genitale interne feminine, dar hipoplazice Absenţa gonadelor, locul lor fiind luat de bandelete alb-sidefii (streak-gonade)
")
76
Examenul citogenetic constată:
Examenul histopatologic al streak-gonadei arată elemente de tromă ovariană turbionată şi lipsită de foliculi Examenul citogenetic constată: Cromatină sexuală absentă Cariotipul 45, X sau mozaicisme cu linii 45, X.

77
SINDROMUL OVARELOR PAUPERE
Este un sindrom caracterizat prin incapacitatea ovarelor de a sexualiza normal datorită unui dispozitiv folicular sexoidogenetic sărac Isuficienta ovariana prematura – denumire actuala a sindomului defineste aceasta entitate patologica, spontana sau familiara, in care femeile isi epuizeaza zestrea foliculara inainte de 40 de ani. Afecteaza aproximativ 1% dintre femei ( procent care intra in menopauza inainte de 40 de ani) In afara de factorul genetic se poate lua in cosiderare etiologia autoimuna, distrugerea ovariana prin radiatii ionizante sau chimioterapice In raport cu gradul pauperităţii ovariene, clinic, insuficienţa ovariană se poate manifesta ca severă sau atenuată
 In afara de factorul genetic se poate lua in cosiderare etiologia autoimuna, distrugerea ovariana prin radiatii ionizante sau chimioterapice. In raport cu gradul pauperităţii ovariene, clinic, insuficienţa ovariană se poate manifesta ca severă sau atenuată.")
78
Evaluarea de laborator a insuficientei ovariene premature
FSH (pentru a stabili diagnosticul de insuficienta ovariana prematura) Cariotipul (<30 ani sau cu infantilism sexual) Testare pentru gena FMR1 (premutation carrier state - FMR1, fragile-X mental retardation 1.) Thyroid-stimulating hormone (hypothyroidism) Tratamentul Substitutiv estro-progesteronic cu doze mici de estrogeni)
 Cariotipul (<30 ani sau cu infantilism sexual) Testare pentru gena FMR1 (premutation carrier state - FMR1, fragile-X mental retardation 1.) Thyroid-stimulating hormone (hypothyroidism) Tratamentul. Substitutiv estro-progesteronic cu doze mici de estrogeni)")
79
Insuficienţa ovariană severă:
Pauperizarea foliculară este atât de intensă, încât ovarele sunt incapabile să iniţieze sexualizarea Morfotipul îmbracă aspect eucunoid Vârsta pubertăţii este atinsă şi depăşită fără a se produce fenomene de feminizare caracteristice: Tegumentele rămân glabre; Sânii nu se dezvoltă – amastie; Menstrele nu apar – amenoree primară;

80
Organele genitale externe sunt feminine, normal conforme sau infantile
Dozările hormonale arată: Estrogenii şi progesteronul au valori practic nule; Gonadotrofinele au valori crescute. Laparotomia exploratorie evidenţiază: Tractul genital intern feminin dar hipoplazic În sit-ul ovarian, ovarele par normale

81
Insuficienţă ovariană atenuată
Cantitatea de foliculi ovarieni este subnormală, ceea ce conferă ovarului o capacitate feminizantă insuficientă şi de scurtă durată. Morfotipul este feminin, dar cu note de hipoovarie

82
Pubertatea întârzie şi este incompletă:
Pilozitatea sexuală se dezvoltă relativ; Sânii au aspect pubertar: areola şi mameloanele mici, slab pigmentate; Menstrele apar târziu, sunt neregulate şi se suspendă după o perioada relativ scurtă – câteva luni sau câţiva ani (menopauza precoce) Organele genitale externe, feminine, sunt normal conformate, dar uneori cu note de hipoplazie
 Organele genitale externe, feminine, sunt normal conformate, dar uneori cu note de hipoplazie.")
83
Dozările hormonale arată, în perioadele de activitate ovariană, valori normale ale estrogenilor şi progesteronului asemănătoare celor constatate la femeia adultă; în perioadele de amenoree, valorile sunt scăzute. Gonadotrofinele sunt normale în perioadele de activitate ovariană şi crescute în perioadele de amenoree

84
Laparotomia exploratorie pune în evidenţă:
Organe genitale interne feminine normale În sit-urile ovariene: ovare aparent normale Examenul histopatologic al ovarelor constă în aspectul normal al ovarelor, dar cu populaţie foliculară relativ redusă: corpi albicans prezenţi, însă rari.

85
Disgenezia monoovariană
Este un sindrom determinat de prezenţa unei singure gonade – ovarul disgenezic Morfotipul este feminin, cu fenomene de sexualizare feminină mai mult sau mai puţin bine dezvoltat- în raport cu capacitatea funcţională a ovarului disgenetic prezent

86
Sexualizarea pubertară:
Uneori lipseşte, situaţia în care morfotipul îmbracă aspect eucunoid, cu pilozitate sexuală absentă sau foarte slab reprezentată: amastie, amenoree primară; Alteori pubertate întârziată, lentă şi incompletă: pilozitate sexuală prezentă, dar insuficient reprezentată, sânii mai mult sau mai puţin bine dezvoltaţi, menstre rare şi suspendate de timpuriu.

87
Organele genitale externe sunt feminine, infantile, hipoplazice sau aparent normal dezvoltate
Laparotomia evidenţiază : Organe genitale externe feminine Ca gonade:un singur ovar, iar de partea controlaterală – o bandeletă sidefie (streak-gonade)
")
88
Dozările hormonale arată valori scăzute sau cvasi normale ale estrogenilor şi progesteronului; gonadotrofinele hipofizare sunt crescute sau normale Examenul histopatologic: Gonada este un ovar disgenetic cu foliculi atrezici, uneori cu corpi albicans şi foliculi evolutivi Streak-gonade: ţesut fibros alcătuit din stromă cu aspect ovarian (turbionată), dar fără foliculi.
, dar fără foliculi.")
89
Defecte in actiunea androgenilor ( sindroame de insensibilitate (rezistenta) partiala sau totala la androgeni), Receptorul androgen este esential pentru medierea actiunilor androgenilor, inculsiv pentru diferentierea organelor genitale externe si interne. Gena receptorului androgen se afla situata pe bratul lung al cromozomului X ( Xq11-q12). Clinic, sunt descrise doua forme ale acestui sindom: forma completa si forma incompleta de insensibilitate la androgeni. Afectiunea relativ rara cu o incidenta de 1 caz la noi nascuti(49). In forma completa (sau sindromul testiculului feminizant, sau al "femeilor fara par", Sdr. Morris), fenotipul indivizilor 46,XY este tipic unul feminin, la care lipsesc atat derivatele wolffiene (insensibilitate la testosteron); lipsesc si derivatele mülleriene ( datorita secretiei de AMH), si lipseste diferentierea organelor genitale externe in sens masculin (insensibilitate la testosteron), acestea fiind tipic feminine . Majoritatea cazurilor sunt datorate unei singure mutatii ( un aminoacid) si sunt mostenite ( ca o afectiune genetica X linkata recesiva, transmisa de mama si exprimata de fatul de sex masculin care mosteneste cromozomul X la care este prezenta mutatia (50).
. Clinic, sunt descrise doua forme ale acestui sindom: forma completa si forma incompleta de insensibilitate la androgeni. Afectiunea relativ rara cu o incidenta de 1 caz la noi nascuti(49). In forma completa (sau sindromul testiculului feminizant, sau al femeilor fara par , Sdr. Morris), fenotipul indivizilor 46,XY este tipic unul feminin, la care lipsesc atat derivatele wolffiene (insensibilitate la testosteron); lipsesc si derivatele mülleriene ( datorita secretiei de AMH), si lipseste diferentierea organelor genitale externe in sens masculin (insensibilitate la testosteron), acestea fiind tipic feminine . Majoritatea cazurilor sunt datorate unei singure mutatii ( un aminoacid) si sunt mostenite ( ca o afectiune genetica X linkata recesiva, transmisa de mama si exprimata de fatul de sex masculin care mosteneste cromozomul X la care este prezenta mutatia (50).")
90
Diagnosticul se stabileste de obicei la pubertate, datorita amenoreei primare ( pacienta nu are uter, vaginul este orb, "in deget de manuse", pilozitatea pubiana si axilara sunt absente. Sanii se dezvolta normal datorita conversiei periferice a testosteronului de catre aromatza. Hormonologic se pune in evidenta valori crescute de LH, FSH si de Testosteron, estradiol crescut pentru un barbat. Provocarea clinicianului o reprezinta dupa diagnosticul genetic, cariotip 46,XY, Barr negativ, absenta uterului, localizarea testiculelor.Acestea se gasesc cel mai frecvent in labiile mari (hernia inghinala este rarisima la fete) dar se pot gasi si intraabdominal, acestea prezentand un risc de degenerare maligna intr-un procent de peste 20% dupa aparitia pubertatii ( sunt specialisti care recomanda castrarea prepubertar pentru a evita malignizarea) (51). Ulterior pacienta va fi tratata cu estrogeni si daca este necesar se va recurge la plastie chirurgicala vaginala. Forma partiala implica un raspuns partial, variabil la actiunea testosteronului cu un grad variat de ambiguitate la nivelul organelor genitale externe ( de la micropenis<2,5cm, hipospadias, criptorhidism, iar derivatele wolffiene sunt bine diferentiate. Castrarea este obligatorie in aceste cazuri, riscul de malignizare fiind de peste 50% pentru cazurile cu criptorhidism ( indiferent de localizarea extrascrotala) (52). . Ulterior se recurge la tratamentul substitutiv hormonal in concordanta cu sexul fenotipic ales.
 dar se pot gasi si intraabdominal, acestea prezentand un risc de degenerare maligna intr-un procent de peste 20% dupa aparitia pubertatii ( sunt specialisti care recomanda castrarea prepubertar pentru a evita malignizarea) (51). Ulterior pacienta va fi tratata cu estrogeni si daca este necesar se va recurge la plastie chirurgicala vaginala. Forma partiala implica un raspuns partial, variabil la actiunea testosteronului cu un grad variat de ambiguitate la nivelul organelor genitale externe ( de la micropenis<2,5cm, hipospadias, criptorhidism, iar derivatele wolffiene sunt bine diferentiate. Castrarea este obligatorie in aceste cazuri, riscul de malignizare fiind de peste 50% pentru cazurile cu criptorhidism ( indiferent de localizarea extrascrotala) (52). . Ulterior se recurge la tratamentul substitutiv hormonal in concordanta cu sexul fenotipic ales.")
92
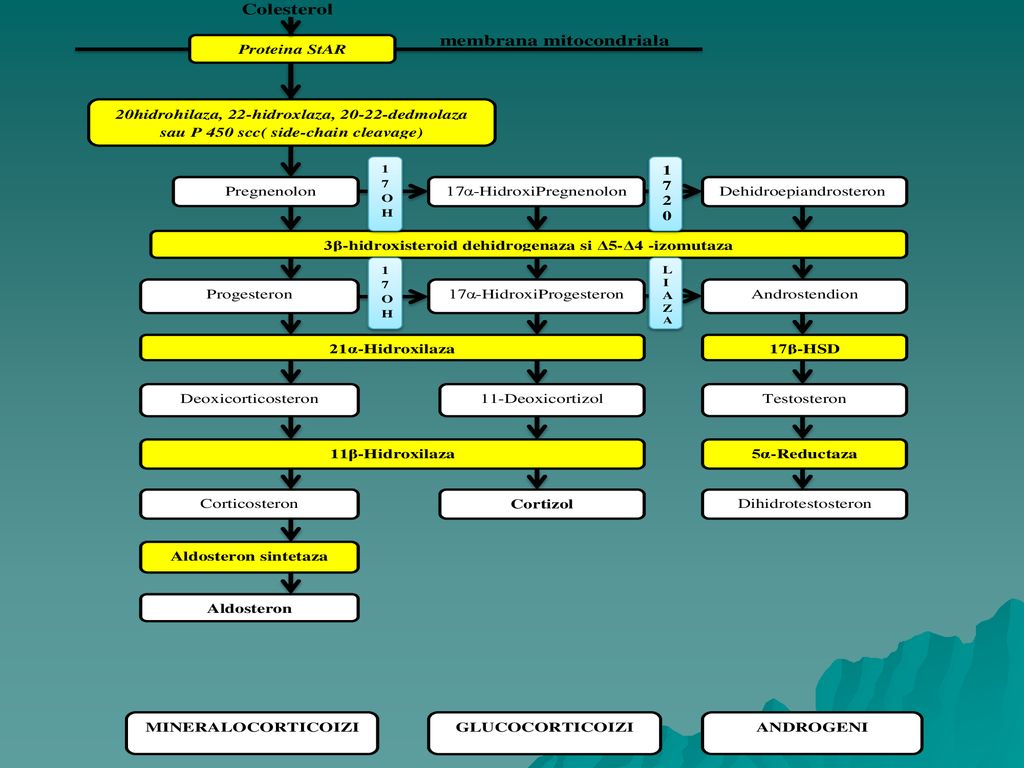
Deficitul de 21-hidroxilaza si de 11βhidroxilaza
Forma congenitala (apare in urma Hiperplaziei congenitale adrenale), iar in functie de enzima implicata(deficit enzimatic), poate antrena atat 46,XY TDS cat si 46,XX TDS. Deficitul de 21-hidroxilaza reprezinta forma cea mai frecventa din punct de vedere etiologic a cazurilor de ambiguitate sexuala la nou nascut. Prin blocarea atat a caii mineral- corticoide cat si a caii glucocorticoide se deturneaza sinteza adrenala spre sinteza prioritara e androgeni care va determina virilizarea fetusilor 46,XX. Incidenta este de 1:15.000, cu variatii. Prezentarea la nastere se face cu ambiguitatea organelor genitale externe si cu un sindrom de pierdere de sare, care in forma completa reprezinta o urgenta. La pacientii 46XY, se exprima clinic printr-o virilizare precoce, statura finala mai mica si infunctie de intensitatea deficitului enzimatic este insotita de un sindrom de pierdere de sare. Confirmarea diagnosticului hormonal se realizeaza prin evidentierea valorilor mult crescute de 17-OH Progesteron.
, iar in functie de enzima implicata(deficit enzimatic), poate antrena atat 46,XY TDS cat si 46,XX TDS. Deficitul de 21-hidroxilaza reprezinta forma cea mai frecventa din punct de vedere etiologic a cazurilor de ambiguitate sexuala la nou nascut. Prin blocarea atat a caii mineral- corticoide cat si a caii glucocorticoide se deturneaza sinteza adrenala spre sinteza prioritara e androgeni care va determina virilizarea fetusilor 46,XX. Incidenta este de 1:15.000, cu variatii. Prezentarea la nastere se face cu ambiguitatea organelor genitale externe si cu un sindrom de pierdere de sare, care in forma completa reprezinta o urgenta. La pacientii 46XY, se exprima clinic printr-o virilizare precoce, statura finala mai mica si infunctie de intensitatea deficitului enzimatic este insotita de un sindrom de pierdere de sare. Confirmarea diagnosticului hormonal se realizeaza prin evidentierea valorilor mult crescute de 17-OH Progesteron.")
93
Deficitul de 11β-hidroxilaza spre deosebire de deficitul descris anterior duce la acumularea de Deoxicorticosteron ( DOC ), cu activitate mineralcorticoida si de androgeni, care explica ambiguitatea organelor genitale externe la nou nascutii 46,XX si hipertensiune arteriala si alcaloza hipokaliemica la ambele sexe. Diagnosticul genetic pune in evidenta modificarea genelor enzimelor deficiente. Abordarea terapeutica a formelor de Hiperplazie congenitala adrenala, ar trebui initiata "in utero" pentru a evita virilizarea fatului de sex feminin.Tratamentul vizeaza supresia adrenostatului cu dexametazona, inainte chiar de determinarea sexului gonadic, unii recomandand administrarea dupa saptamana a 4 a de sarcina.. La fetusii 46,XX afectati de gena se recomanda tratamentul pe tot parcursul sarcinii, pana la nastere. Dupa nastere tratamentul substitutiv glucocorticoid (hidrocortizon), este obligatoriu pentru prevenirea insuficientei adrenale acute, indiferent de forma de deficit enzimatic.
, este obligatoriu pentru prevenirea insuficientei adrenale acute, indiferent de forma de deficit enzimatic.")
Παρόμοιες παρουσιάσεις