Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
Bolile genetice Boli ereditare Mostenite fie sub aceeasi forma
fie ca susceptibilitate, predispozitie

2
Tipuri de boli ereditare
1. Bolile cromozomiale Monosomii (45, X; 5p-) Trisomii (47XXY; sindroamele Down, Edwards, Patau) 2. Bolile monogenice Autozomale: - dominante - recesive Gonozomale: 3. Bolile multifactoriale (boli cardiace, autoimune) 4. Bolile mitocondriale 5. Boli cauzate de mutatii dinamice
 Trisomii (47XXY; sindroamele Down, Edwards, Patau) 2. Bolile monogenice. Autozomale: - dominante. - recesive. Gonozomale: 3. Bolile multifactoriale (boli cardiace, autoimune) 4. Bolile mitocondriale. 5. Boli cauzate de mutatii dinamice.")
3
CURIOSI?

4
Bolile genetice erau considerate raritati, intalnite intamplator de catre medici evidentierea lor este o urmare: a diminuarii frecventei bolilor ce nu au origine genetica si a dezvoltarii tehnologiei necesare studiului cromozomial si genic/molecular impactul asupra sanatatii populatiei nu este nesemnificativ se cunosc peste de boli conditionate genetic Afecteaza cel putin 5 – 8% dintre nou-nascuti (nn)
")
5
Bolile genetice Au devenit o importanta cauza de morbiditate si mortalitate Sunt boli cronice Genereaza handicap fizic si/sau psihic Sunt o problema majora de sanatate publica Impun actiuni concrete si eficace de diagnostic (dg.), un program national de profilaxie, bazat pe sfat genetic, screening pre- si neonatal, registre nationale si dg. presimptomatic in familiile cu risc crescut
, un program national de profilaxie, bazat pe sfat genetic, screening pre- si neonatal, registre nationale si dg. presimptomatic in familiile cu risc crescut.")
6
Genetica medicala este o parte a geneticii umane
este in Romania o specialitate clinica distincta se ocupa de: dg. si ingrijirea pacientilor cu boli genetice familile pacientilor anual aproximativ nn vor fi afectati de boli genetice inainte de varsta de 25 ani in Romania acest fapt a necesitat crearea unei retele nationale de Centre de Genetica Medicala Regionale si Cabinete Judetene

7
BOLILE MONOGENICE SUNT AFECTIUNI PRODUSE DE MODIFICARI (MUTATII) ALE UNEI SINGURE GENE SE TRANSMIT IN SUCCESIUNEA GENERATIILOR RESPECTA TRANSMITEREA MENDELIANA FIIND DE ACEEA NUMITE SI BOLI MENDELIENE ONLINE MENDELIAN INHERITANCE IN MAN (OMIM) initiat de regretatul prof. Victor McKusick cuprinde circa de intrari (gene) si explica evolutia cercetarilor corelatiei dintre fenotip si genotip se estimeaza a fi peste 4000 de boli genetice cauzate de defecte monogenice Genomic imprinting si disomia uniparentala schimba modul de transmitere Amplificarile genice anormale = mut. Dinamic Iulie 2008 la 86 a murit Mc Kusick
 initiat de regretatul prof. Victor McKusick cuprinde circa de intrari (gene) si explica evolutia cercetarilor corelatiei dintre fenotip si genotip. se estimeaza a fi peste 4000 de boli genetice cauzate de defecte monogenice. Genomic imprinting si disomia uniparentala schimba modul de transmitere. Amplificarile genice anormale = mut. Dinamic. Iulie 2008 la 86 a murit Mc Kusick.")
8
BOLILE MONOGENICE Sunt numite boli moleculare, daca se cunosc mutatia genica si proteina pe care aceasta o codifica Majoritatea devin evidente clinic deja din perioada neonatala sau din copilarie (ex.fenilcetonuria, mucoviscidoza) Dupa pubertate debuteaza clinic doar aprox. 10% dintre bolile mongenice cunoscute (ex.distrofia musculara Becker) 1% devin manifeste clinic dupa sfarsitul perioadei reproductive (ex. boala Huntington) Desi rare, frecventa lor globala la copii este de aprox. 2 – 3% Era medicinii moleculare
 Dupa pubertate debuteaza clinic doar aprox. 10% dintre bolile mongenice cunoscute (ex.distrofia musculara Becker) 1% devin manifeste clinic dupa sfarsitul perioadei reproductive (ex. boala Huntington) Desi rare, frecventa lor globala la copii este de aprox. 2 – 3% Era medicinii moleculare.")
9
In continuare numai cateva dintre
bolile monogenice

10
PREVALENTA UNOR BOLI MONOGENICE
HIPERCOLESTEROLEMIA FAMILIALA (AD) 1 la 500 nou-nascuti ANEMIA FALCIFORMA (AR) 1 la 650 nn afroamericani RINICHIUL POLICHISTIC (AD) 1 la nn MUCOVISCIDOZA/FIBROZA CHISTICA (AR) 1 la nn caucazieni COREEA HUNTINGTON (AD) 1 la nn DISTROFIA MUSCULARA DUCHENNE/BECKER (XR) 1 la nn HEMOFILIA (XR) 1 la nn FENILCETONURIA (AR) 1 la nn SINDROMUL MARFAN (AD) 1 la nn
 1 la 500 nou-nascuti. ANEMIA FALCIFORMA (AR) 1 la 650 nn afroamericani. RINICHIUL POLICHISTIC (AD) 1 la nn. MUCOVISCIDOZA/FIBROZA CHISTICA (AR) 1 la nn caucazieni. COREEA HUNTINGTON (AD) 1 la nn. DISTROFIA MUSCULARA DUCHENNE/BECKER (XR) 1 la nn. HEMOFILIA (XR) 1 la nn. FENILCETONURIA (AR) 1 la nn. SINDROMUL MARFAN (AD) 1 la nn.")
11
Caracteristicile bolilor autozomal recesive
Ele se manifesta la persoanele homozigote pentru mutatia cauzatoare a patologiei De obicei persoana afectata are parinti sanatosi clinic, dar heterozigoti, purtatori ai mutatiei Un cuplu format din heterozigoti pentru una si aceeasi mutatie, are un risc de 25% de a avea un copil afectat de boala produsa de acea mutatie Transmisia nu este pe verticala, caci sare generatii, dar este pe “orizontala”, putandu-se manifesta la mai multe persoane intr-o generatie

12
Genotipul descendentilor Fenotipul descendentilor
Parinti Genotipul descendentilor Fenotipul descendentilor 1) AA x AA 100% AA (homozygotes) 100% A 2) AA x Aa 50% AA ; 50% Aa (homo-; heterozygotes) 3) AA x aa 100% Aa (heterozygotes) 4) Aa x aa 50% Aa; 50% aa (homo-; heterozygotes) 50% A; 50% a 5) Aa x Aa 25% AA; 50% Aa; 25% aa (homo-; heterozygotes) 75% A; 25% a 6) aa x aa 100% aa (homozygotes) 100% a
 AA x AA. 100% AA (homozygotes) 100% A. 2) AA x Aa. 50% AA ; 50% Aa (homo-; heterozygotes) 3) AA x aa. 100% Aa (heterozygotes) 4) Aa x aa. 50% Aa; 50% aa (homo-; heterozygotes) 50% A; 50% a. 5) Aa x Aa. 25% AA; 50% Aa; 25% aa. (homo-; heterozygotes) 75% A; 25% a. 6) aa x aa. 100% aa (homozygotes) 100% a.")
13
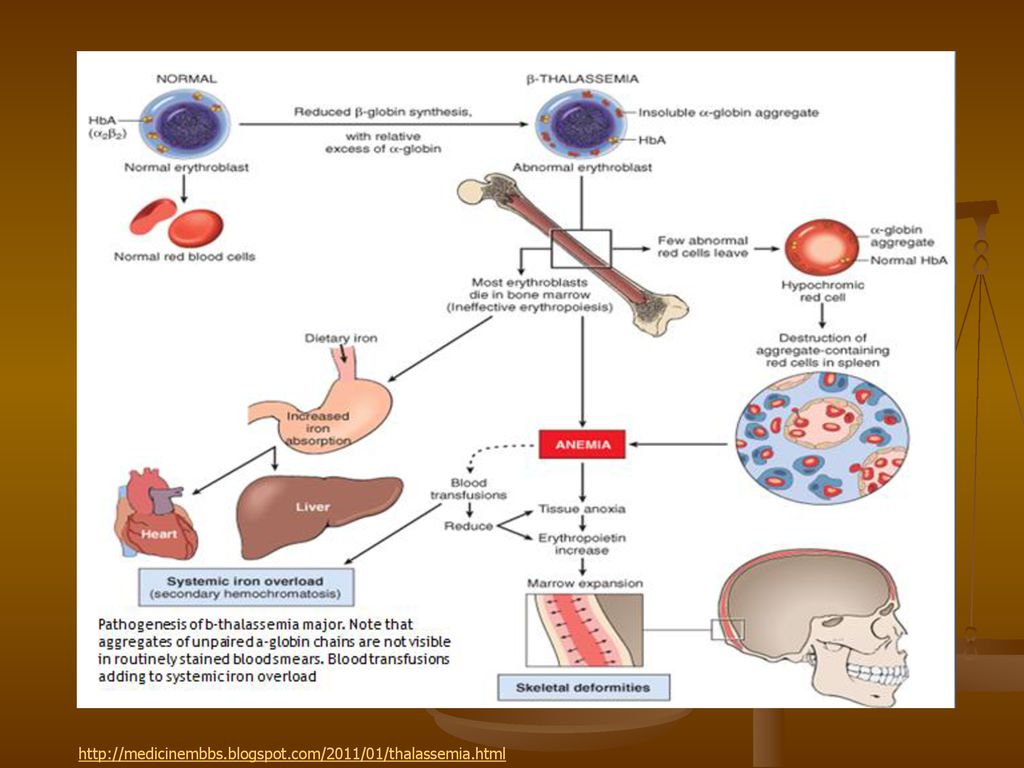
Hemoglobinopatiile reprezentate in principal de talazemii si anemia falciforma (drepanocitoza, sicklemia – ‘sickle- cell anaemia’) sunt raspandite pe tot globul aproape 5% din populatia globului este purtatoare de gene responsabile de aparitia hemoglobinopatiilor in fiecare an aproape de copii se nasc cu hemoglobinopatii, dintre care peste sunt bolnavii din Africa cu drepanocitoza mondial sunt insa mai multi purtatori de modificari generatoare de talazemii decat de drepanocitoza prezenta mutatiei punctiforme ( A T = transversie) in codonul 6 al genei ß-globinei este insa foarte mare in anumite zone, ceea ce are drept urmare o rata crescuta a drepanocitozei comparativ cu talazemiile (prin casatorii se intalnesc mai frecv. heterozigotii)
 in codonul 6 al genei ß-globinei este insa foarte mare in anumite zone, ceea ce are drept urmare o rata crescuta a drepanocitozei comparativ cu talazemiile (prin casatorii se intalnesc mai frecv. heterozigotii)")
14
Anemia falciforma De exemplu, in Nigeria 24% din populatie poarta mutatia, iar prevalenta anemiei este de aproape 20 la 1000 nn. Ceea ce inseamna ca numai in Nigeria, se nasc anual aproape de copii bolnavi! Boala este prezenta mai ales in Africa Sahariana, India, Arabia Saudita si tarile din jurul Mediteranei Initial s-a crezut ca originea mutatiei este in Peninsula Araba, de unde s-ar fi raspandit catre Asia si Africa. Astazi se stie, ca au fost cel putin 4 evenimente mutationale diferite, 3 in Africa si al patrulea fie in Arabia Saudita fie in centrul Indiei. Aceste evenimente independente au avut loc in urma cu de generatii, adica cu aproximativ de ani. Distributia reflecta faptul ca boala confera avantaj selectiv impotriva malariei, cauzata de Plasmodium (malariae, falciparum, etc) Vivax, ovale
 Vivax, ovale.")
15
Moleculele de Hb din eritrocite transporta oxigen catre toate tesuturile.
Hb (S) = hemoglobina anormala, ce cristalizeaza in interiorul hematiei conferindu-i forma de secera, ea pierzand capacitatea de a se deforma pentru a strabate capilarele sanguine; aa 6 al β-globinei (ac. glutamic) este inlocuit de valina Aceste celule in forma de secera sunt fragile, nu pot transporta suficient O2 si nu pot traversa peretele capilarelor, pentru a ajunge la tesuturi.... producand dureri (osoase,viscerale), anemie, stare generala alterata, ajungand fara tratament chiar la deces. Ocluzia capilarelor de catre hematiile anormale conduce la microinfarcte Hemoliza este urmata de splenomegalie si scaderea imunitatii infectii bacteriene (a se vedea cursul 9!)
 = hemoglobina anormala, ce cristalizeaza in interiorul hematiei conferindu-i forma de secera, ea pierzand capacitatea de a se deforma pentru a strabate capilarele sanguine; aa 6 al β-globinei (ac. glutamic) este inlocuit de valina. Aceste celule in forma de secera sunt fragile, nu pot transporta suficient O2 si nu pot traversa peretele capilarelor, pentru a ajunge la tesuturi.... producand dureri (osoase,viscerale), anemie, stare generala alterata, ajungand fara tratament chiar la deces. Ocluzia capilarelor de catre hematiile anormale conduce la microinfarcte. Hemoliza este urmata de splenomegalie si scaderea imunitatii infectii bacteriene. (a se vedea cursul 9!)")
16
RFLP-uri produse din fragmente ale genei pt beta globina
Diagnostic molecular Folosind enzime de restrictie (endonucleaze) RFLP-uri produse din fragmente ale genei pt beta globina RFLP = restriction fragment length polimorphism /polimorfismul lungimii fragmentului de restrictie
 RFLP-uri produse din fragmente ale genei pt beta globina. RFLP = restriction fragment length polimorphism /polimorfismul lungimii. fragmentului de restrictie.")
17
Diagnostic prenatal si sfat genetic pentru cupluri la risc
Tratamentul Analgezice in crizele vasculo-ocluzive dureroase Penicilina, alte antibiotice mai ales pana la varsta de 5 ani Acid folic zilnic Transfuzii Transplant medular Alte tratamente: Hidroxiuree, cianat Terapia genica: speranta de a reactiva genele in vederea sintezei HbF, cu afinitate mare pentru oxigen Diagnostic prenatal si sfat genetic pentru cupluri la risc

18
http://thalassemianme. webs

19
Talazemiile Hemoglobinopatii cauzate de mutatii ale genelor β si α globinei, de pe cz.16 si respectiv 11, ce au ca urmare sinteza deficitara a lantului polipeptidic corespunzator Predomina in jurul Mediteranei, in Tailanda, Malaezia, Filipine si Africa Exista 3 mecanisme de aparitie a α – talazemiilor; Deletii mari al ambelor gene α (α1 si α 2) Crossing-over inegal Foarte rar mutatii: fie de tip nonsens (apare un codon ‘stop’ pozitionat anormal), fie de tip ‘frameshift’ (decaleaza cadrul de lectura)
 Crossing-over inegal. Foarte rar mutatii: fie de tip nonsens (apare un codon ‘stop’ pozitionat anormal), fie de tip ‘frameshift’ (decaleaza cadrul de lectura)")
20
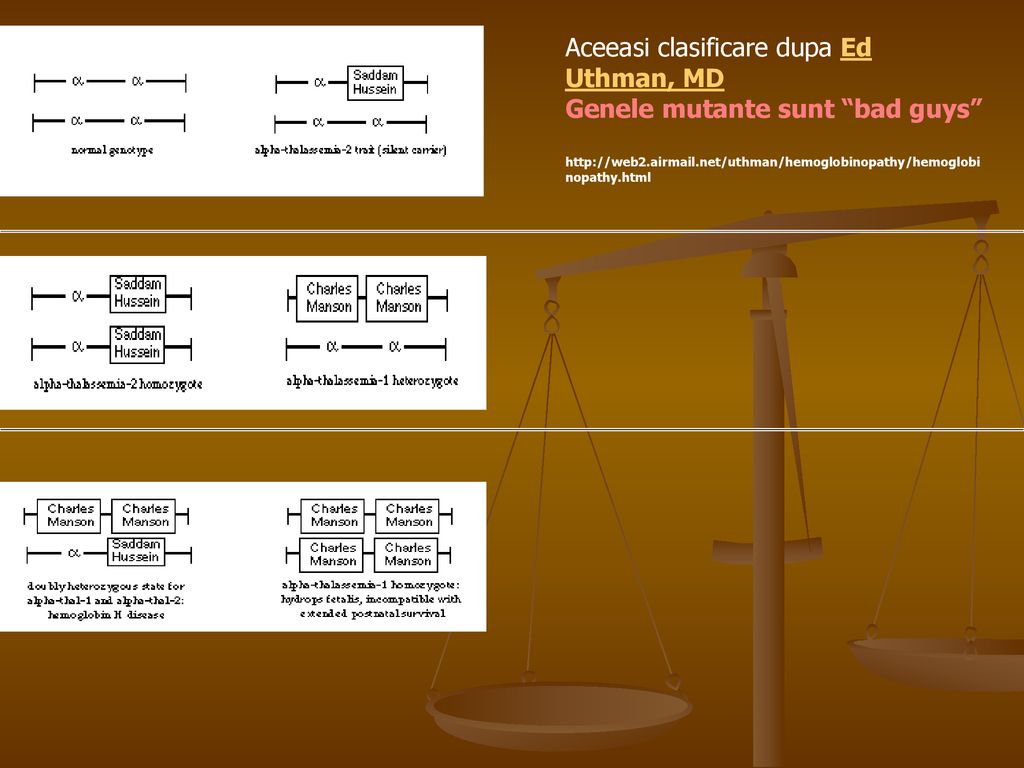
Relatia dintre severitatea talazemiei (fenotip) si numarul de gene α nefunctionale (genotip)
genotip (cz. 16/ cz. 16) normal α1 α2 / α1 α2 Purtator “silentios”; htz. α2 α1 - / α1 α2 Caracter α – talazemic; htz. α2 si α1 - - / α1 α2 Caracter α – talazemic; homoz. α2 α1 - / α1 - Hemoglobinoza H (Hb H = β4) Anemie hemolitica; neletala α1 - / - - Hidrops fetal (Hb Bart = 4 lanturi gamma) - - / - -
 normal. α1 α2 / α1 α2. Purtator silentios ; htz. α2. α1 - / α1 α2. Caracter α – talazemic; htz. α2 si α / α1 α2. Caracter α – talazemic; homoz. α2. α1 - / α1 - Hemoglobinoza H (Hb H = β4) Anemie hemolitica; neletala. α1 - / - - Hidrops fetal (Hb Bart = 4 lanturi gamma) - - / - -")
21
Aceeasi clasificare dupa Ed Uthman, MD Genele mutante sunt “bad guys”
22
α – talazemia Deletia dublu homozigota conduce la deces perinatal:
- FAT NASCUT MORT - DECES NEONATAL PRECOCE Celelalte forme nu sunt grave, deoarece se mai sintetizeaza Hb A In cazul hidropsului fetal, Hb fetala majoritara este un tetramer al lanturilor γ numit Hb Bart, pe cand in cazul hemoglobinozei tetramerul prezent la adulti este β4 Hb Bart are capacitate extrem de redusa de transport a oxigenului, astfel tesuturile fetale sunt putin oxigenate prin oxigenul dizolvat in sangele periferic. Asociaza o anemie severa. Pompa miocardica, deja deficitar oxigenata, utilizeaza un surplus de energie pentru a face sa circule putinul oxigen dizolvat in sangele periferic; acest travaliu conduce la o insuficienta cardiaca, ce se manifesta prin edeme importante (“hidrops fetal”), ce sunt incompatibile cu supravietuirea.
, ce sunt incompatibile cu supravietuirea.")
23
HIDROPS FETAL

24
β- Talazemiile Spre deosebire de cele α, care sunt mai ales produse prin deletii, acestea apar in urma mutatiilor punctiforme Intre 1982 – 1992 s-au studiat si descoperit peste 90 de mutatii diferite, ce pot determina β – talazemii, demonstrand heterogenitatea alelica (diferite mutatii produc acelasi fenotip) prezenta in acest caz. De fapt, majoritatea pacientilor cu β – talazemie majora sunt heterozigoti compusi, mostenind de la fiecare parinte cate o alela β – talazemica purtatoare a unei mutatii diferite. Mutatiile sunt dispersate pe toata lungimea genei β – globinei, inclusiv la nivelul promotorului (exceptie face ‘cutia’ CCAAT)
 prezenta in acest caz. De fapt, majoritatea pacientilor cu β – talazemie majora sunt heterozigoti compusi, mostenind de la fiecare parinte cate o alela β – talazemica purtatoare a unei mutatii diferite. Mutatiile sunt dispersate pe toata lungimea genei β – globinei, inclusiv la nivelul promotorului (exceptie face ‘cutia’ CCAAT)")
25
Tipuri de mutatii in beta-talazemii:
Mutatii la nivelul promotorului (substitutii) precum: Prima cutie CACCC are ultima CT A doua cutie CACCC are a doua CT si a treia C G In cutia ATAA apare substitutia A G Mutatii (in principal la nivelul exonilor) de tip ‘nonsens’ (TAA, TAG,TGA pozitionati anormal) ‘frame-shift’ (decaleaza cadrul de lectura) Ele se produc precoce in secventa ADN a.i.mai mult de ½ din structura proteica este absenta, conducand la un produs genic nefunctional, extrem de instabil, ce este degradat rapid in hematii. Persoanele homozigote pt aceasta mutatie au β0- talazemie Mutatii la nivelul situsurilor de clivare GT si AG conduc tot la β0- talazemii In tarile mediteraniene exista o mutatie in intronul 1 pozitia 110, G A; astfel apare un nou situs acceptor de matisare (AG), ARNm matur va contine 19 nucleotide suplimentare (≠ de multiplu de 3) producandu-se un decalaj al cadrului de lectura. Surprinzator, desi in 90% din cazuri este luata in considerare pozitia cu mutatia, raman 10% din ARNpm care vor citi corect a.i. un individ chiar homozigot pt aceasta mutatie, va avea un fenotip β+- talazemic, deoarece se poate detecta o cantitate redusa de β- globina.
 precum: Prima cutie CACCC are ultima CT. A doua cutie CACCC are a doua CT si a treia C G. In cutia ATAA apare substitutia A G. Mutatii (in principal la nivelul exonilor) de tip. ‘nonsens’ (TAA, TAG,TGA pozitionati anormal) ‘frame-shift’ (decaleaza cadrul de lectura) Ele se produc precoce in secventa ADN a.i.mai mult de ½ din structura proteica este absenta, conducand la un produs genic nefunctional, extrem de instabil, ce este degradat rapid in hematii. Persoanele homozigote pt aceasta mutatie au β0- talazemie. Mutatii la nivelul situsurilor de clivare GT si AG conduc tot la β0- talazemii. In tarile mediteraniene exista o mutatie in intronul 1 pozitia 110, G A; astfel apare un nou situs acceptor de matisare (AG), ARNm matur va contine 19 nucleotide suplimentare (≠ de multiplu de 3) producandu-se un decalaj al cadrului de lectura. Surprinzator, desi in 90% din cazuri este luata in considerare pozitia cu mutatia, raman 10% din ARNpm care vor citi corect a.i. un individ chiar homozigot pt aceasta mutatie, va avea un fenotip β+- talazemic, deoarece se poate detecta o cantitate redusa de β- globina.")
26
In talazemia intermediara: fie ambele gene ale β – globinei sunt anormale, fie una este anormala si cealalta lipseste

27
Beta talazemia majora (anemia Cooley)
Boala nu se evidentiaza la nastere, ci progresiv in cursul primului an de viata apare o anemie severa, urmare a scaderii Hb F si respectiv deficitului din ce in ce mai mare de β – globina. Ca raspuns la aceasta anemie, maduva osoasa – in care hematiile sunt distruse, adica inainte ca ele sa ajunga in sange – este sediul unei eritropoieze care este intensa dar ineficienta. Ficatul si splina se maresc (hiperplazie); Hipotrofie staturo-ponderala, anomalii scheletice si fragilitate osoasa (oase subitri), facies usor mongoloid Analizele de laborator arata o anemie hemolitica, hipocroma cu sideremie crescuta, poliglobulie si microcitoza Ficatul si splina = sedii ale unei eritropoieze extramedulare Gr. Haima, haimatos = sange, iar gr. lysis = dizolvare, distructie hemoliza = eritrocitoliza = fenomen de distrugere a globulelor rosii cu eliberarea Hb
; Hipotrofie staturo-ponderala, anomalii scheletice si fragilitate osoasa (oase subitri), facies usor mongoloid. Analizele de laborator arata o anemie hemolitica, hipocroma cu sideremie crescuta, poliglobulie si microcitoza. Ficatul si splina = sedii ale unei eritropoieze extramedulare. Gr. Haima, haimatos = sange, iar gr. lysis = dizolvare, distructie hemoliza = eritrocitoliza = fenomen de distrugere a globulelor rosii cu eliberarea Hb.")
28
Modificari craniene (bose) datorita expansiunii maduvei rosii hematogene la acest nivel
Icter: o colorare galbena a pielii si sclerei

29
TABLOUL CLINIC – ANEMIA COOLEY
SPLENOMEGALIE ANEMIE (nr. redus de hematii) HEPATOMEGALIE ICTER MODIFICARI OSOASE (fracturi frecvente) Rx: hiperplazia maduvei osoase la nivelul craniului MODIFICARI ALE FORMEI SI MARIMII FETEI SI CRANIULUI
31
Beta talazemia majora (anemia Cooley)
Fara tratament decesul survine inainte de varsta de 10 ani, cu alterarea starii generale, paloare, lipsa poftei de mancare, cu anemie severa si infectii grave. Complicatiile ce conduc la deces sunt: ciroza, insuficienta hepatica si cea cardiaca. Cu tratament se poate atinge varsta adulta

32
TRATAMENT TRANSFUZII TRANSPLANT MEDULAR TERAPIE CU CHELATORI AI FIERULUI (Deferoxamine) Acid Folic Splenectomie Terapie genica in vederea stimularii sintezei de gamma globina SFAT GENETIC

33
Tratamentul β -talazemiei
Tratamentul conventional: Transfuzii (risc in timp, de acumulare a Fe ce se depune in diferite organe: inima, ficat, pancreas) Deferoxamina (Desferal) – chelator al Fe, favorizeaza excretia urinara a acestuia Tratamente posibile: Grefa medulara (din 1981, dar nr. mic de pacienti gasesc donor corespunzator; procedura in sine este si ea riscanta) Transplant de celule stem din cordonul ombilical
 Deferoxamina (Desferal) – chelator al Fe, favorizeaza excretia urinara a acestuia. Tratamente posibile: Grefa medulara (din 1981, dar nr. mic de pacienti gasesc donor corespunzator; procedura in sine este si ea riscanta) Transplant de celule stem din cordonul ombilical.")
34
Splenectomia evidentiaza o splina giganta de 1 kg
Splenomegalie; Splenectomia evidentiaza o splina giganta de 1 kg
35
Dar trata- mentul nu este lipsit de complicatii Rareori ajung
la varsta adulta
36
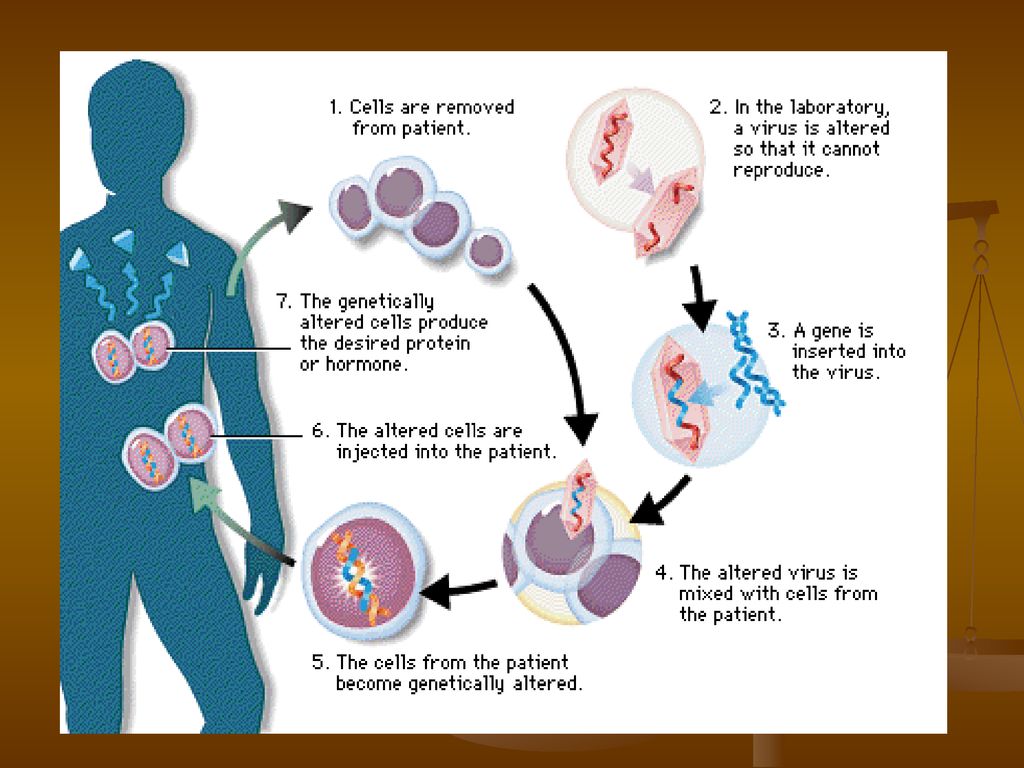
Tratamentul β -talazemiei
Tratamentul genic (speranta pt. viitor): Transferul unei gene normale de β – globina in celulele stem, imature ale maduvei osoase, care sunt precursorii tuturor celorlalte celule sanguine. O alta forma de terapie genica ar putea implica folosirea unor medicamente sau a altor metode de reactivare a genelor γ – globinei in vederea producerii de Hb F – ceea ce ar compensa deficitul de Hb A al pacientilor
: Transferul unei gene normale de β – globina in celulele stem, imature ale maduvei osoase, care sunt precursorii tuturor celorlalte celule sanguine. O alta forma de terapie genica ar putea implica folosirea unor medicamente sau a altor metode de reactivare a genelor γ – globinei in vederea producerii de Hb F – ceea ce ar compensa deficitul de Hb A al pacientilor.")
Παρόμοιες παρουσιάσεις