Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
Ευθυγράμμιση πολλαπλών ακολουθιών

2
Σκοπός Ευθυγράμμισης πολλών ακολουθιών
Αποκαλύπτει περιοχές πρωτεϊνών που διατηρούνται κοινός πρόγονος. Συνέπεια του ανωτέρω μακρινές (εξελικτικά) σχέσεις μεταξύ πρωτεϊνών εντοπίζονται ευκολότερα.
 σχέσεις μεταξύ πρωτεϊνών εντοπίζονται ευκολότερα.")
3
Παράδειγμα Π.χ. ακολουθίες industry interesting important Ευθυγράμμιση
Αυτό δεν είναι ευθυγράμμιση (γιατί;) in-du--stry- intere-sting im-por--tant
 in-du--stry- intere-sting. im-por--tant.")
4
Αλγόριθμοι ευθυγράμμισης πολλών ακολουθιών
Δυναμικός προγραμματισμός Σταδιακές μέθοδοι (Progressive Methods) Επαναληπτικές μέθοδοι (Iterative Methods)
 Επαναληπτικές μέθοδοι (Iterative Methods)")
5
Παράδειγμα με δυναμικό προγραμματισμό

6
Ευθυγράμμιση πολλών ακολουθιών.
Απαιτείται χρόνος Ο(2^k*n^k) για k ακολουθίες μεγέθους n Aπαιτείται χώρος O(n^k) Πχ. 5 ακολουθίες μέσου μήκους 250, απαιτείται Χώρος: 975 Gbytes Βήματα: 31Χ1012 Ενώ για 2 ακολουθίες μέσου μήκους 250, απαιτείται Χώρος: 60kbytes Βήματα:240X103 Συνεπώς χρήσιμος ο αλγόριθμος για μικρές ακολουθίες και για λίγες.
 για k ακολουθίες μεγέθους n. Aπαιτείται χώρος O(n^k) Πχ. 5 ακολουθίες μέσου μήκους 250, απαιτείται. Χώρος: 975 Gbytes. Βήματα: 31Χ1012. Ενώ για 2 ακολουθίες μέσου μήκους 250, απαιτείται. Χώρος: 60kbytes. Βήματα:240X103. Συνεπώς χρήσιμος ο αλγόριθμος για μικρές ακολουθίες και για λίγες.")
7
Σταδιακές μέθοδοι βέλτιστης ευθυγράμμισης (progressive methods of msa)
Η μέθοδος Δυναμικού προγραμματισμού περιορίζεται σε μικρές ακολουθίες ή σε λίγες .... Σταδιακές μέθοδοι: ξεκίνα από τις κοντινότερες ακολουθίες και σταδιακά βάζε τις μακρινότερες.

8
Παράδειγμα σταδιακής ευθυγράμμισης
Π.χ. NYLS, NKYLS, NFS, NFLS NYLS NKYLS NFS NFLS +K -L N F L /- S Y to F N K /- Y/F L /-S

9
Παράδειγμα σταδιακής ευθυγράμμισης
Π.χ. NYLS, NKYLS, NFS, NFLS NYLS NKYLS NFS NFLS N K / - Y L S N F L /- S seqA: N _ F L S seqB: N _ F _ S seqC: N K Y L S seqD: N _ Y L S N K /- Y/F L /-S

10
Εμπορικά προγράμματα σταδιακής ευθυγράμμισης
CLUSTALW ( PILEUP

11
Clustal W Ευθυγράμμιση ακολουθιών (ανά δύο).
Κατασκευή φυλογενετικού δέντρου, βάσει των scores. Ευθυγράμμιση βάσει φυλογενετικού δέντρου.

12
Πως προκύπτει το δένδρο;
Συγκρίνω τις ακολουθίες ανά δύο Έστω s1,s2,s3,s4,s5 Με τον ακόλουθο πίνακα (ευθυγραμμίσεις ανά δύο) Έστω μικροί αριθμοί δηλώνουν μεγάλη ομοιότητα s1 s2 s3 s4 s5 2 6 10 9 5 8 4
 Έστω μικροί αριθμοί δηλώνουν μεγάλη ομοιότητα. s1. s2. s3. s4. s")
13
Πως προκύπτει το δένδρο;
Κοντινότερες s1,s2 τις ομαδοποιώ s1,s2 s3 s4 s5 5 9 8 4 3

14
Πως προκύπτει το δένδρο;
Ομαδοποιώ τις s4,s5 s1,s2 s3 s4,s5 5 8 4

15
Πως προκύπτει το δένδρο;
Ομαδοποιώ τις (s4,s5), s3 s1,s2 s3,(s4,s5) 5 s3, (s4,s5)
, s3. s1,s2. s3,(s4,s5) 5. s3, (s4,s5)")
16
Δέντρο

17
Βαθμολόγηση ευθυγράμμισης, μέθοδος SP, βαθμολογώ ανά 2, και παίρνω το άθροισμα
IN-DU-STRY- INTERESTING IM-POR-TANT IN-DU-STRY- INTERESTING Σκορ 3 INTERESTING IM-POR-TANT Σκορ 5 IN-DU-STRY- IM-POR-TANT Σκορ 7 15

18
Προβλήματα με την προοδευτική ευθυγράμμιση
Η τελική ευθυγράμμιση εξαρτάται από τις αρχικές ευθυγραμμίσεις ζευγών Αν οι αρχικές ευθυγραμμίσεις δεν είναι καλές, θα προκύψει MSA με χαμηλό σκορ Σημαντικό ρόλο παίζουν και οι πίνακες αντικατάστασης.

19
Επαναληπτικές μέθοδοι ευθυγράμμισης (Iterative Methods of MSA)
Διαρκής επαναευθυγράμμιση υποσυνόλων των ακολουθιών Στόχος: Βελτίωση του συνολικού σκορ ευθυγράμμισης Μέθοδος: Γενετικοί αλγόριθμοι.

20
Γενετικοί αλγόριθμοι α’
Μηχανική μάθηση Ενισχυτική μάθηση Βασισμένος στην θεωρία της εξέλιξης. Δημιουργία πολλών δυνατών MSAs Δηλαδή Πολλές διαφορετικές θέσεις εισαγωγής κενών Πολλές διαφορετικές αντικαταστάσεις. Π.χ.

21
Γενετικοί αλγόριθμοι β’
Αρχικοποίηση αλγορίθμου: δημιουργία πολλών τυχαίων ευθυγραμμίσεων. Αποτιμάται κάθε μία με τη μέθοδο του αθροίσματος των διαφορών. Οι περισσότερες δεν είναι καλές. Βήμα 2: Οι μισές καλύτερες ακολουθίες πάνε στην επόμενη γενιά. Οι χειρότερες πάνε ανάλογα με την βαθμολογία τους. Υποβάλλονται σε μετάλλαξη Όλες υποβάλλονται σε επανασυνδυασμό για την επόμενη γενιά

22
Γενετικοί αλγόριθμοι δ’, μεταλλάξεις
XXXXXXXX XXX---XXX—XX XXXXXXXX X—XXX---XXXX Δηλαδή στις μεταλλάξεις βάζω σε τυχαίες θέσεις κενά, με μικρή πιθανότητα

23
Γενετικοί αλγόριθμοι δ’, επανασυνδυασμός
Γονέας Γονέας 2 M G K V N - - V D E - G E A L - Μ Α Κ V Ν V A D – D E - G E A L M K K V G - - D H A – - G E A L - M G K V N - - V D E G E A L Μ Α Κ V Ν V A D – D E G E A L M K K V G D H A G E A L Παιδί 1 Παιδί 2 M G K V - N - - V D E G E A L Μ Α Κ V Ν V A D – D E G E A L M K K V G D H A G E A L M G K V N - - V D E - G E A L - Μ Α Κ V - Ν V A D – D E - G E A L M K K V - G - - D H A – - G E A L

24
Τοπικές Ευθυγραμμίσεις Πολλών ακολουθιών (Local msa)
Μέθοδοι : Ανάλυση profiles Ανάλυση των blocks Αναζήτηση προτύπων,pattern searching (eMotif)
")
25
Profiles, γιατί; Μεγαλύτερη ακρίβεια στις ευθυγραμμίσεις
Εντοπίζονται ευκολότερα άλλες ομόλογες ακολουθίες Τα α/α που έχουν χαμηλό βαθμό διατήρησης πιθανώς περιοχές που έλκουν αντισώματα σχεδιασμός αντιβιοτικών.

26
Παράδειγμα profile O πίνακας έχει 23 στήλες 20 για τα 20 αμινοξέα
1 στήλη για ένα άγνωστο αμινοξύ z 2 στήλες για εισαγωγή κενού και ποινή προέκτασης του.

27
Παράδειγμα profile Υπάρχει μία γραμμή για κάθε στήλη της msa.
Οι τιμές κάθε γραμμής δείχνουν τον αριθμό εμφάνισης κάθε αμινοξέως στις ακολουθίες. Για παράδειγμα στην πρώτη γραμμή τα αμινοξέα I,T,V παρουσιάστηκαν με το I να έχει την πλειοψηφία εμφανίσεων. Αν θέλουμε να ψάξουμε για ακολουθία μήκους π.χ. 100, και έχω profile 10, χρησιμοποιώ παράθυρο. Η μεγαλύτερη θετική τιμή κάθε στήλης είναι η στήλη που αντιστοιχεί στο αμινοξύ της consensus ακολουθίας Υπάρχουν δύο μέθοδοι κατασκευής profile: Average και Evolutionary

28
Μέθοδος κατασκευής Average
Π.χ. Αν στην στήλη 1 της MSA έχει 5 Ι 3 Τ 2 V (ισοπίθανα) 0.5, 0.3, 0.2 συχνότητες Ι-Ι, Ι-Τ, Ι-V (Pam250) 0.5x5+0.3x0+0.2x4=3.3
 0.5, 0.3, 0.2 συχνότητες. Ι-Ι, Ι-Τ, Ι-V (Pam250) 0.5x5+0.3x0+0.2x4=3.3.")
29
Χρήση πίνακα profile Ψάξε βάσεις δεδομένων για ακολουθίες πρωτεϊνών με το ίδιο pattern Χρησιμοποίησέ το ως πίνακα ταιριάσματος σε msa.

30
Ανάλυση των blocks Αντιπροσωπεύουν μια διατηρημένη περιοχή της msa.
Διαφέρουν από τα profiles στο ότι δεν έχουν κενά παρά μόνο θέσεις που ταυτίζονται ή που δεν ταυτίζονται. Blocks μπορούν να εξαχθούν από μια Ευθυγράμμιση Πολλών ακολουθιών(msa) χρησιμοποιώντας τον αλγόριθμο BLOCKS
 χρησιμοποιώντας τον αλγόριθμο BLOCKS.")
31
Blocks Blocks database: www.blocks.fhcrc.org
Προέρχονται από την βάση interpro

32
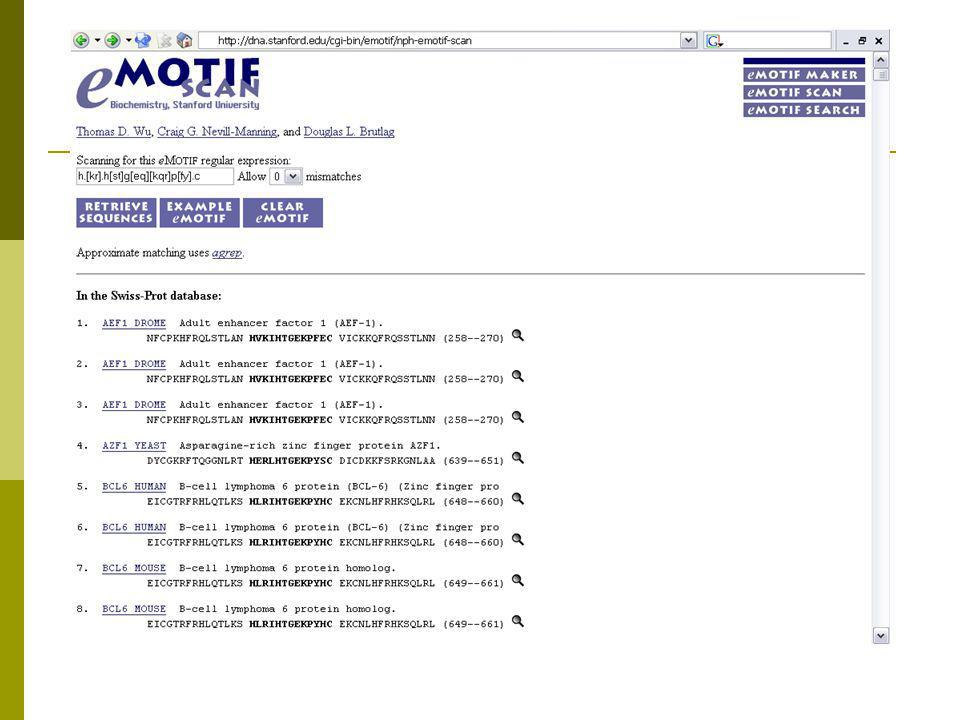
Αναζήτηση προτύπων(eMOTIF)
Έχουν βρεθεί από τις βάσεις Δεδομένων BLOCKS και HSSP σετ ομάδων αντικατάστασης αμινοξέων για κάθε στήλη Ευθυγραμμίσων Πολλών Ακολουθιών. Στο msa μιας ομάδας πρωτεϊνών, εξετάζεται κάθε στήλη για να δούμε αν αυτά τα σετ που έχουν βρεθεί βρίσκονται στη στήλη της ευθυγράμμισης. Ακολουθεί παράδειγμα :

33
Αναζήτηση προτύπων (eMOTIF)
Δύο ομάδες, βάσει R/L στην 4η θέση, και Y στο τέλος Στήλη 1, έχουμε την ομάδα Μ γιατί το Μ υπάρχει πάντα στη στήλη. Στήλη 2, το Y και το F του σετ FYW βρέθηκε σε αυτή τη στήλη άρα το σετ FYW χρησιμοποιείται για αυτή τη στήλη κ.ο.κ FWY: aromatic group

34
eMOTIF P(motif)=p(M)x[p(F)+p(W)+p(y)]x[p(k)+p(r))
P(i), συχνότητες των αμινοξέων στην SwissProt Εκτίμηση της ποιότητας του motif eMOTIF δημιουργεί πολλά motifs Επιλέγουμε τα πλέον ευαίσθητα (p(motif)) Δηλαδή αυτά που βρίσκουν πολλές ακολουθίες. Identify: βάση motifs dna.stanford.edu/emotif
![eMOTIF P(motif)=p(M)x[p(F)+p(W)+p(y)]x[p(k)+p(r))](http://slideplayer.gr/slide/2311378/8/images/34/eMOTIF+P%28motif%29%3Dp%28M%29x%5Bp%28F%29%2Bp%28W%29%2Bp%28y%29%5Dx%5Bp%28k%29%2Bp%28r%29%29.jpg "P(i), συχνότητες των αμινοξέων στην SwissProt. Εκτίμηση της ποιότητας του motif. eMOTIF δημιουργεί πολλά motifs. Επιλέγουμε τα πλέον ευαίσθητα (p(motif)) Δηλαδή αυτά που βρίσκουν πολλές ακολουθίες. Identify: βάση motifs dna.stanford.edu/emotif.")
36
Hidden Markov Models α HMM στατιστικό μοντέλο για ευθυγράμμιση ακολουθιών. Αρχικοποίηση μοντέλο κατασκευάζεται με εκτιμήσεις για τις ακολουθίες που έχουμε Στη συνέχεια «εκπαίδευση» του μοντέλου με ακολουθίες Χρήση μοντέλου Παραγωγή της καλύτερης MSA Έρευνα για εύρεση παρόμοιων ακολουθιών

37
Hidden Markov Models β Πρακτικά
Το ίδιο καλή (ως προς MSA) με άλλες μεθόδους. Βάσεις σε θεωρία πιθανοτήτων Δεν απαιτείται ειδική διάταξη των ακολουθιών που ευθυγραμμίζουμε Sequence Alignment and Modeling System (SAM)
 με άλλες μεθόδους. Βάσεις σε θεωρία πιθανοτήτων. Δεν απαιτείται ειδική διάταξη των ακολουθιών που ευθυγραμμίζουμε. Sequence Alignment and Modeling System (SAM)")
38
Hidden Markov Models γ Είσοδος: Ο χρήστης παρέχει την πρωτεΐνη στόχο ή ένα καλό MSA Έξοδος: το μοντέλο O ρόμβος αντιστοιχεί στη εισαγωγή αμινοξέως/βάσης, ο κύκλος στην διαγραφή αμινοξέως/βάσης το τετράγωνο στο ταίριασμα

39
Hidden Markov Models δ

40
Hidden Markov Models P(TAC)=0.7*0.5*0.7*0.4*0.7*0.4*0.9=0.025
=0.7*0.5*0.7*0.4*0.7*0.4*0.9=0.025")
41
Ηidden markov models P(TAC)=0.025
Μετατροπή της πιθανότητας σε log odds (δες αποτέλεσμα σε επόμενη διαφάνεια Για κάθε βάση ισχύει log2 (p(βάσης) /(1/αριθμό βάσεων), π.χ. Για το πρώτο τετράγωνο για Α, -1.32=log2(0.1/0.25) Για τις μεταβάσεις log2(p(μετάβασης)/(1/αριθμό μεταβάσεων), π.χ. Για την πρώτη μετάβαση -1.72=log2(0.7/0.333) Αρα το log odds score για την ακολουθία TAC είναι: > =6.4800 Ίδια λογική και με το score ευθυγράμμισης πρωτεϊνών
 /(1/αριθμό βάσεων), π.χ. Για το πρώτο τετράγωνο για Α, -1.32=log2(0.1/0.25) Για τις μεταβάσεις log2(p(μετάβασης)/(1/αριθμό μεταβάσεων), π.χ. Για την πρώτη μετάβαση -1.72=log2(0.7/0.333) Αρα το log odds score για την ακολουθία TAC είναι: > = Ίδια λογική και με το score ευθυγράμμισης πρωτεϊνών.")
42
Hidden markov model with log odd scores

43
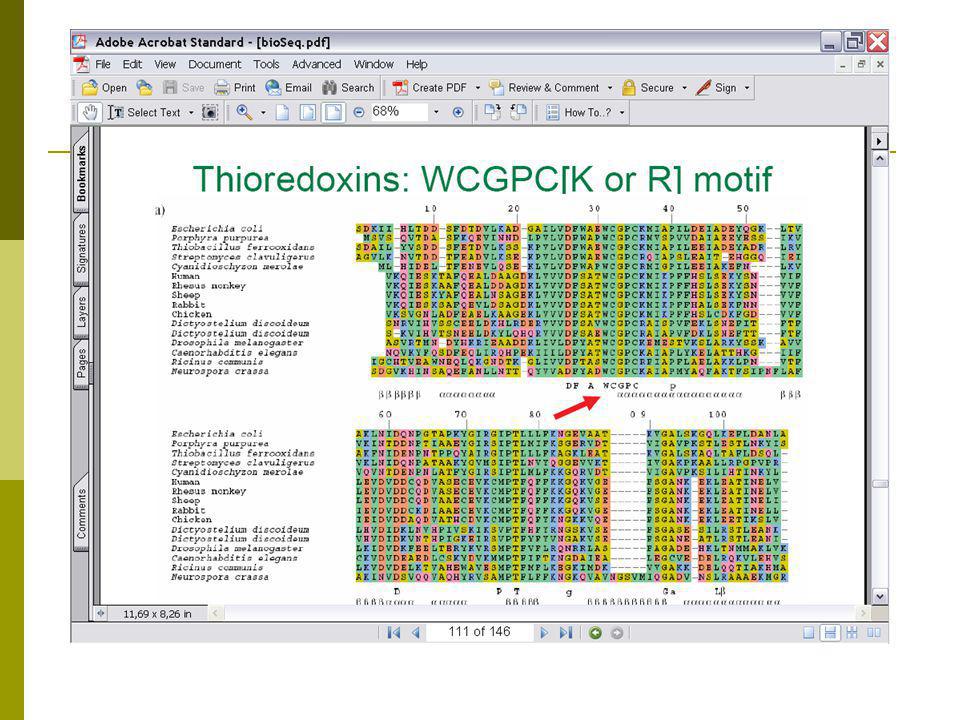
Συμπεράσματα από τη χρήση ευθυγραμμίσεων για πρόβλεψη δομής
Οι Thierodoxines είναι ένζυμα, (η εικόνα είναι από το E.coli).
.")
44
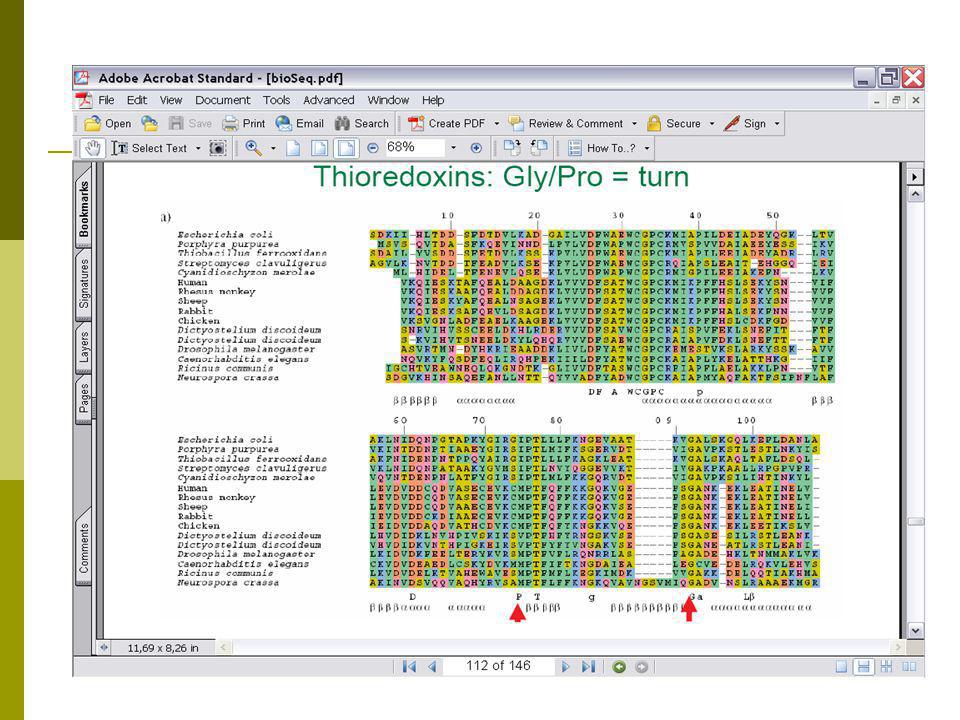
Συμπεράσματα από τη χρήση ευθυγραμμίσεων για τη δομή β’
Οι περισσότερο διατηρημένες περιοχές αντιστοιχούν στην ενεργό περιοχή. H «γέφυρα» στις περιοχές 32 και 35 του E.coli. Περιοχές με πολλές εισαγωγές και διαγραφές αντιστοιχούν σε surface loops. Περιοχές με διατήρηση Gly ή Pro αντιστοιχούν σε “turn” Turns 9,20,60,95 Διατηρημένη περιοχή με υδρόφοβα α/α υποδεικνύει έλικα (40-49)
")
47
Χρωματισμός/ομαδοποίηση Αμινοξέων
Χρώμα Τύπος Αμινοξέα Κίτρινο Μικρά μη πολικά Gly (G), Ala (A), Ser (S), Thr (T) Πράσινο Υδρόφοβα Cys (C), Val (V), Ile (I), Leu (L), Pro (P), Phe (F), Tyr (Y), Met (M), Trp (W) Ιώδες Πολωμένα Asn (N) , Gln (Q), His (H) Κόκκινο Αρνητικό φορτίο Asp (D) , Glu (E) Μπλέ Θετικό φορτίο Lys (K), Arg (R)
, Ala (A), Ser (S), Thr (T) Πράσινο. Υδρόφοβα. Cys (C), Val (V), Ile (I), Leu (L), Pro (P), Phe (F), Tyr (Y), Met (M), Trp (W) Ιώδες. Πολωμένα. Asn (N) , Gln (Q), His (H) Κόκκινο. Αρνητικό φορτίο. Asp (D) , Glu (E) Μπλέ. Θετικό φορτίο. Lys (K), Arg (R)")
Παρόμοιες παρουσιάσεις



















=α, κκπ(τ,σ)=ν, κκπ(λ,π)=η κκπ(π,σ)=γ, κκπ(ξ,ο)=κ ξο κκπ(ι,ξ)=β, κκπ(τ,θ)=θ, κκπ(ο,μ)=α.>")