Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
Εισαγωγή στη Βιοπληροφορική
Αθανάσιος Τσακαλίδης, Χρήστος Μακρής Επικουρικό: Ξενοφών Ευαγγελόπουλος Μάθημα Ελεύθερης Επιλογής Τομέα Λογικού των Υπολογιστών Εαρινό Εξάμηνο Ακαδ. Έτους Εισαγωγή στη Βιοπληροφορική Μακρής

2
Τεχνικές Ανάλυσης και Σύγκρισης Ακολουθιών Βιολογικών Δεδομένων
Προσεγγιστική Εύρεση Προτύπου - Approximate Pattern Matching Στοίχιση Ακολουθιών - Multiple Sequence Alignment Εφαρμογές σε Προβλήματα Μοριακής Βιολογίας

3
Βασικοί Ορισμοί (α) Απόσταση Μετασχηματισμού - Edit Distance: για 2 συμβολοσειρές ορίζουμε το ελάχιστο πλήθος των πράξεων μετασχηματισμού που απαιτούνται για να μετασχηματίσουμε την πρώτη συμβολοσειρά στη δεύτερη. Οι βασικές πράξεις μετασχηματισμού είναι η ένθεση, διαγραφή και αντικατάσταση συμβόλων. Παράδειγμα: S1: vintner και S2: writers edit-distance(S1->S2)=5 Λέγεται και Levenshtein distance, παραμένει το ίδιο είτε αν η ακολουθία πράξεων εφαρμόζεται στο S1 είτε στο S2.
=5. Λέγεται και Levenshtein distance, παραμένει το ίδιο είτε αν η ακολουθία πράξεων εφαρμόζεται στο S1 είτε στο S2.")
4
Βασικοί Ορισμοί (β) ένθεση: I διαγραφή: D αντικατάσταση: R
Ακολουθία Μετασχηματισμού - Edit Transcript: για το μετασχηματισμό μιας συμβολοσειράς ορίζεται ως η ακολουθία των πράξεων μετασχηματισμού που απαιτούνται για να μετασχηματίσουμε την πρώτη συμβολοσειρά στη δεύτερη. Οι βασικές πράξεις μετασχηματισμού αναπαρίστανται ως εξής: ένθεση: I διαγραφή: D αντικατάσταση: R ταίριασμα: Μ Παράδειγμα: S1: vintner και S2: writers edit-distance(S1->S2)= RIMDMDMMI
= RIMDMDMMI.")
5
Στοίχιση Ακολουθιών Στοίχιση Ακολουθιών- Sequence Alignment: τοποθετούμε τη μια ακολουθία κάτω από την άλλη έτσι ώστε οι κοινοί χαρακτήρες να τοποθετούνται στις ίδιες θέσεις.

6
Στοίχιση Ακολουθιών επιτρέποντας κενά
Στοίχιση δυο ακολουθιών με την εισαγωγή 7 κενών χαρακτήρων σε 4 θέσεις, που μεταφράζεται ως μετάλλαξη της ακολουθίας του DNA στις αντίστοιχες θέσεις.

7
Η Μέθοδος του Δυναμικού Προγραμματισμού
Δυναμικός Προγραμματισμός: Έστω 2 ακολουθίες S1 και S2, θα συμβολίζουμε ως D(i,j) την απόσταση μετασχηματισμού μεταξύ των προθεμάτων S1[1..i] και S2[1..j], δηλαδή τον ελάχιστο αριθμό πράξεων μετασχηματισμού που απαιτούνται για να μετασχηματίσουμε τους i πρώτους χαρακτήρες της ακολουθίας S1 στους j πρώτους χαρακτήρες της ακολουθίας S2. Χρήση 3 βασικών τεχνικών: σχέση αναδρομής- recurrence relation, χρήση πίνακα- tabular computation, σχέση οπισθοχώρησης- traceback.
 την απόσταση μετασχηματισμού μεταξύ των προθεμάτων S1[1..i] και S2[1..j], δηλαδή τον ελάχιστο αριθμό πράξεων μετασχηματισμού που απαιτούνται για να μετασχηματίσουμε τους i πρώτους χαρακτήρες της ακολουθίας S1 στους j πρώτους χαρακτήρες της ακολουθίας S2. Χρήση 3 βασικών τεχνικών: σχέση αναδρομής- recurrence relation, χρήση πίνακα- tabular computation, σχέση οπισθοχώρησης- traceback.")
8
Παράδειγμα Πίνακα Δυναμικού Πρ/σμου

9
D(i,j)=min[D(i-1,j)+1,D(i,j-1)+1,D(i-1,j-1)+t(i,j)]
Η Σχέση Αναδρομής Σχέση Αναδρομής: D(i,j)=min[D(i-1,j)+1,D(i,j-1)+1,D(i-1,j-1)+t(i,j)] D(i,j-1)+1: πρέπει να ενθέσουμε το χαρακτήρα S2[j] D(i-1,j)+1: πρέπει να διαγράψουμε το χαρακτήρα S2[j], D(i-1,j-1)+1: για να μετασχηματίσουμε το χαρακτήρα S1[i] στο χαρακτήρα S2[j] πρέπει να αντικαταστήσουμε το χαρακτήρα S1[i], με το χαρακτήρα S2[j], D(i-1,j-1): έχουμε ταίριασμα
![D(i,j)=min[D(i-1,j)+1,D(i,j-1)+1,D(i-1,j-1)+t(i,j)]](http://slideplayer.gr/slide/2032663/8/images/9/D%28i%2Cj%29%3Dmin%5BD%28i-1%2Cj%29%2B1%2CD%28i%2Cj-1%29%2B1%2CD%28i-1%2Cj-1%29%2Bt%28i%2Cj%29%5D.jpg "Η Σχέση Αναδρομής. Σχέση Αναδρομής: D(i,j)=min[D(i-1,j)+1,D(i,j-1)+1,D(i-1,j-1)+t(i,j)] D(i,j-1)+1: πρέπει να ενθέσουμε το χαρακτήρα S2[j] D(i-1,j)+1: πρέπει να διαγράψουμε το χαρακτήρα S2[j], D(i-1,j-1)+1: για να μετασχηματίσουμε το χαρακτήρα S1[i] στο χαρακτήρα S2[j] πρέπει να αντικαταστήσουμε το χαρακτήρα S1[i], με το χαρακτήρα S2[j], D(i-1,j-1): έχουμε ταίριασμα.")
10
Παράδειγμα: σχέση αναδρομής

11
Η Σχέση Οπισθοχώρησης Σχέση Οπισθοχώρησης:
από την (i,j) θέση προς την (i,j-1) αν D(i,j)= D(i,j-1)+1 (ένθεση χαρακτήρα) από την (i,j) θέση προς την (i-1,j) αν D(i,j)= D(i-1,j)+1 (διαγραφή χαρακτήρα) από την (i,j) θέση προς την (i-1,j-1) αν D(i,j)= D(i-1,j- 1)+t(i,j) (αντικατάσταση χαρακτήρα ή ταίριασμα)
 θέση προς την (i,j-1) αν D(i,j)= D(i,j-1)+1 (ένθεση χαρακτήρα) από την (i,j) θέση προς την (i-1,j) αν D(i,j)= D(i-1,j)+1 (διαγραφή χαρακτήρα) από την (i,j) θέση προς την (i-1,j-1) αν D(i,j)= D(i-1,j- 1)+t(i,j) (αντικατάσταση χαρακτήρα ή ταίριασμα)")
12
Προσθήκη δεικτών οπισθοχώρησης

13
Ερμηνεία δεικτών οπισθοχώρησης

14
Πολυπλοκότητα της μεθόδου Δυναμικού Προγραμματισμού
Αρχικοποίηση: O(n) + O(m) Σχέση Αναδρομής: O(n*m) Δείκτες Οπισθοχώρησης: Ο(n+m) Πολυπλοκότητα: Ο(n2) Ισοδυναμία με πρόβλημα της θεωρίας γραφημάτων, όπου κάθε κόμβος έχει ετικέτα ένα ζεύγος (i,j)
 + O(m) Σχέση Αναδρομής: O(n*m) Δείκτες Οπισθοχώρησης: Ο(n+m) Πολυπλοκότητα: Ο(n2) Ισοδυναμία με πρόβλημα της θεωρίας γραφημάτων, όπου κάθε κόμβος έχει ετικέτα ένα ζεύγος (i,j)")
15
Βασικοί Ορισμοί (γ) ένθεση ή διαγραφή: d αντικατάσταση: r
Ζυγισμένη Απόσταση Μετασχηματισμού - Weighted Edit Distance: το ελάχιστο κόστος των πράξεων μετασχηματισμού που απαιτούνται για να μετασχηματίσουμε την πρώτη συμβολοσειρά στη δεύτερη. Κάθε πράξη μετασχηματισμού έχει συγκεκριμένο κόστος - βάρος. Έστω ότι οι βασικές πράξεις μετασχηματισμού έχουν τα ακόλουθα βάρη: ένθεση ή διαγραφή: d αντικατάσταση: r ταίριασμα: m. Παράδειγμα: S1: vintner και S2: writers weighted edit-distance(S1->S2)= r+4d+4m.
= r+4d+4m.")
16
Η Σχέση Αναδρομής με βάρη
Σχέση Αναδρομής: D(i,j)=min[D(i-1,j)+d,D(i,j-1)+d,D(i-1,j-1)+t(i,j)], όπου: t(i,j)= e, αν S1(i)=S2(j), t(i,j)=r, αν S1(i)S2(j) και D(i,0)=i*d και D(0,j)=j*d. Η σχέση μπορεί να υπολογιστεί κινούμενοι κατά στήλες από αριστερά προς τα δεξιά, αφού έχουμε υπολογίσει τη πρώτη γραμμή και τη πρώτη στήλη.
=min[D(i-1,j)+d,D(i,j-1)+d,D(i-1,j-1)+t(i,j)], όπου: t(i,j)= e, αν S1(i)=S2(j), t(i,j)=r, αν S1(i)S2(j) και. D(i,0)=i*d και D(0,j)=j*d. Η σχέση μπορεί να υπολογιστεί κινούμενοι κατά στήλες από αριστερά προς τα δεξιά, αφού έχουμε υπολογίσει τη πρώτη γραμμή και τη πρώτη στήλη.")
17
ΕΠΕΚΤΑΣΕΙΣ Ζυγισμένη Απόσταση Μετασχηματισμού βάσει Αλφαβήτου - Weighted Edit Distance. Σε εφαρμογές Μοριακής Βιολογίας τα βάρη αντικατάστασης χαρακτήρων αποθηκεύονται σε Πίνακες Αντικατάστασης - Substitution Matrix: PAM και BLOSUM Καλύτερη (σημασιολογικά) η αντιμετώπιση σαν alignment και η ενσωμάτωση του score. Κατά κανόνα match είναι θετικό, οτιδήποτε άλλο 0 ή αρνητικό
 η αντιμετώπιση σαν alignment και η ενσωμάτωση του score. Κατά κανόνα match είναι θετικό, οτιδήποτε άλλο 0 ή αρνητικό.")
18
Παράδειγμα Πίνακα Αντικατάστασης
Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής

19
Υπολογισμός score-function στοίχισης ακολουθιών

20
Δυναμικός Προγραμματισμός & Ομοιότητα Ακολουθιών βάσει αλφαβήτου
Σχέση Αναδρομής για την ομοιότητα ακολουθιών: V(i,j)= max[V(i-1,j-1)+s(S1(i), S2(j)), V(i-1,j)+ s(S1(i),_), V(i,j-1)+ s(_,S2(j))], όπου: s(x,y): η τιμή στοίχισης του χαρακτήρα x με τον y V(0,j)=s(_,S2(k)), 1<k<j και V(i,0)= s(S1(k),_), 1<k<i.
= max[V(i-1,j-1)+s(S1(i), S2(j)), V(i-1,j)+ s(S1(i),_), V(i,j-1)+ s(_,S2(j))], όπου: s(x,y): η τιμή στοίχισης του χαρακτήρα x με τον y. V(0,j)=s(_,S2(k)), 1<k<j και V(i,0)= s(S1(k),_), 1<k<i.")
21
Επεκτάσεις Longest Common Subsequence (match weight 1, otherwise 0)
End-space free variant that encourages one string to align in the interior of the other, or the suffix of one to align with the prefix of the other (initial conditions 0, everything is countable in the last row and the last column) – shotgun sequence assembly Approximate occurrence of P in T (the optimal alignment of P to a substring of T has distance δ from the optimal alignment) (initial conditions, as previously 0). -- locate a cell (n,j) with value greater than δ. -- traverse backpointers from (n,j) to (0,k). -- occurrence in T[k,j]
 – shotgun sequence assembly. Approximate occurrence of P in T (the optimal alignment of P to a substring of T has distance δ from the optimal alignment) (initial conditions, as previously 0). -- locate a cell (n,j) with value greater than δ. -- traverse backpointers from (n,j) to (0,k). -- occurrence in T[k,j]")
22
Το Πρόβλημα Τοπικής Στοίχισης Επιθέματος- Local Suffix Alignment Problem
Local suffix alignment problem: για δυο ακολουθίες S1 και S2 εντόπισε ένα επίθεμα α του S1[1..i] (με την πιθανότητα να είναι κενό) και ένα επίθεμα β του S2[1..j] (πιθανόν κενό) τέτοια ώστε το V(α,β) να έχει τη μέγιστη τιμή από όλα τα άλλα δυνατά ζεύγη επιθεμάτων των S1[1..i] και S2[1..j]. Συμβολίζουμε ως υ(i,j) τη βέλτιστη τοπική στοίχιση επιθεμάτων για τις τιμές i και ,j ( i<n και j<m). Αρχικές συνθήκες v(i,0)=0 και v(0,j)=0 καθώς μπορούμε να επιλέξουμε κάθε άδειο επίθεμα. υ(i,j)=max[0,υ(i-1,j-1)+s(S1(i), S2(j)), υ(i-1,j)+ s(S1(i),_), υ(i,j-1)+ s(_,S2(j))]
 και ένα επίθεμα β του S2[1..j] (πιθανόν κενό) τέτοια ώστε το V(α,β) να έχει τη μέγιστη τιμή από όλα τα άλλα δυνατά ζεύγη επιθεμάτων των S1[1..i] και S2[1..j]. Συμβολίζουμε ως υ(i,j) τη βέλτιστη τοπική στοίχιση επιθεμάτων για τις τιμές i και ,j ( i<n και j<m). Αρχικές συνθήκες v(i,0)=0 και v(0,j)=0 καθώς μπορούμε να επιλέξουμε κάθε άδειο επίθεμα. υ(i,j)=max[0,υ(i-1,j-1)+s(S1(i), S2(j)), υ(i-1,j)+ s(S1(i),_), υ(i,j-1)+ s(_,S2(j))]")
23
Locally Similar Strings

24
Παρατηρήσεις Global Alignment is called Needleman-Wunsch alignment
Local alignment is called Smith-Waterman alignment Smith-Waterman can find regions with high similarity by simply performing a trace-back from any cell (i,j) backwards and locate a pair with similarity v(i,j).
 backwards and locate a pair with similarity v(i,j).")
25
Στοίχιση Ακολουθιών με κενά
Έννοια κενού: συνεχόμενα spaces, θέλουμε να ελέγχουμε την κατανομή των κενών. Εισαγωγή Κενών: Για να συμπεριλάβουμε το κόστος που η εισαγωγή κενών εισάγει στη στοίχιση 2 ακολουθιών, μπορούμε σε μια απλή προσέγγιση να θεωρήσουμε ότι κάθε κενό συνεισφέρει ένα σταθερό βάρος Wg, ανεξάρτητα από το μήκος του. τιμή στοίχισης που περιέχει “k” κενά: μία καλύτερη προσέγγιση είναι η χρησιμοποίηση μίας συνάρτησης του μήκους του κενού. Τότε μπορούμε να γεμίσουμε ένα πίνακα: V(i,j)=max[E(i,j), F(i,j), G(i,j)]
=max[E(i,j), F(i,j), G(i,j)]")
26
Στοίχιση με arbitrary gap weights
j G(i,j)= V(i-1,j-1)+cost(i->j) E(i,j)= maxKV(i,k) - w(j-k) (0<=k<=j-1) i gaps j F(i,j)= maxl{V(l,j) - w(i-l) (0<=l<=i-1) i gaps j
= V(i-1,j-1)+cost(i->j) E(i,j)= maxKV(i,k) - w(j-k) (0<=k<=j-1) i. gaps. j. F(i,j)= maxl{V(l,j) - w(i-l) (0<=l<=i-1) i. gaps. j.")
27
V(i,j)=max{E(i,j), F(i,j), G(i,j)}
V(i,0)=-w(i) V(0,j)=-w(j) E(i,0)=-w(i) F(0,j)=-w(j) G(0,0)=0 Assuming that |S1|=n and |S2|=m the recurrences can be evaluated in O(nm2+n2m)
=-w(i) V(0,j)=-w(j) E(i,0)=-w(i) F(0,j)=-w(j) G(0,0)=0. Assuming that |S1|=n and |S2|=m the recurrences can be evaluated in O(nm2+n2m)")
28
Εφαρμογές σε Προβλήματα Μοριακής Βιολογίας
Το Πρόβλημα της Πολλαπλής Στοίχισης- multiple sequence alignment problem: Μία πολλαπλή ολική στοίχιση από k>2 συμβολοσειρές S={ S1, S2,…., Sκ} είναι μία φυσική γενίκευση της στοίχισης για δύο συμβολοσειρές.

29
Γιατί μας ενδιαφέρει η πολλαπλή στοίχιση ακολουθιών
Η πολλαπλή στοίχιση ακολουθιών χρησιμοποιείται: στην αναγνώριση και αναπαράσταση πρωτεϊνικών οικογενειών και υπερ-οικογενειών, στην αναπαράσταση των χαρακτηριστικών που μεταφέρονται στις ακολουθίες DNA ή στις πρωτεϊνικές ακολουθίες, στην αναπαράσταση της εξελικτικής ιστορίας (φυλογενετικά δέντρα) από ακολουθίες DNA ή πρωτεϊνών.
 από ακολουθίες DNA ή πρωτεϊνών.")
30
Αλγόριθμοι Πολλαπλής Στοίχισης Ακολουθιών
Είδη στοίχισης: Extention of DP approach (too costly) Use of pairwise alignment (center star algorithm) Αλγόριθμοι πολλαπλής στοίχισης ακολουθιών: FASTA BLAST.
 Use of pairwise alignment (center star algorithm) Αλγόριθμοι πολλαπλής στοίχισης ακολουθιών: FASTA. BLAST.")
31
Βιολογικές Βάσεις Δεδομένων
Γενικευμένες (Generalised) ή Αρχειακές (Archival) βιολογικές βάσεις δεδομένων). Διακρίνονται σε: - Πρωτογενείς βάσεις δεδομένων ακολουθιών (Primary Sequence Databases). Περιέχουν νουκλεοτιδικές και αμινοξικές ακολουθίες από γονιδιώματα οργανισμών που είτε έχουν αποκρυπτογραφηθεί πλήρως είτε όχι - βάσεις δεδομένων που περιέχουν τρισδιάστατες δομές νουκλεϊνικών οξέων και πρωτεϊνών Δευτερευουσες (Secondary) βιολογικές βάσεις δεδομένων που προκύπτουν από ανάλυση των δεδομένων που είναι αποθηκευμένα στις αρχειακές βιολογικές βάσεις δεδομένων και διακρίνονται σε: Δευτερεύουσες ΒΔ ακολουθιών DNA και πρωτεϊνών που προκύπτουν από τις βασικές ΒΔ ακολουθιών και περιλαμβάνουν (α) ΒΔ ακολουθιών στις οποίες έχουν απομακρυνθεί οι ακολουθίες που έχουν αποθηκευτεί περισσότερες από μία φορές (β) ΒΔ που καταγράφουν μεταλλαγές ή παραλλαγές στις ακολουθίες DNA και πρωτεινών (γ) Γονιδιωματικές ΒΔ που είτε ομαδοποιούν συγγενή ή όχι πλήρως αποκρυπτογραφημένα γονιδιώματα είτε ασχολούνται με γονιδιώματα οργανισμών μοντέλων
 ή Αρχειακές (Archival) βιολογικές βάσεις δεδομένων). Διακρίνονται σε: - Πρωτογενείς βάσεις δεδομένων ακολουθιών (Primary Sequence Databases). Περιέχουν νουκλεοτιδικές και αμινοξικές ακολουθίες από γονιδιώματα οργανισμών που είτε έχουν αποκρυπτογραφηθεί πλήρως είτε όχι. - βάσεις δεδομένων που περιέχουν τρισδιάστατες δομές νουκλεϊνικών οξέων και πρωτεϊνών. Δευτερευουσες (Secondary) βιολογικές βάσεις δεδομένων που προκύπτουν από ανάλυση των δεδομένων που είναι αποθηκευμένα στις αρχειακές βιολογικές βάσεις δεδομένων και διακρίνονται σε: Δευτερεύουσες ΒΔ ακολουθιών DNA και πρωτεϊνών που προκύπτουν από τις βασικές ΒΔ ακολουθιών και περιλαμβάνουν. (α) ΒΔ ακολουθιών στις οποίες έχουν απομακρυνθεί οι ακολουθίες που έχουν αποθηκευτεί περισσότερες από μία φορές. (β) ΒΔ που καταγράφουν μεταλλαγές ή παραλλαγές στις ακολουθίες DNA και πρωτεινών. (γ) Γονιδιωματικές ΒΔ που είτε ομαδοποιούν συγγενή ή όχι πλήρως αποκρυπτογραφημένα γονιδιώματα είτε ασχολούνται με γονιδιώματα οργανισμών μοντέλων.")
32
ΒΔ που ασχολούνται με τις ιεραρχήσεις ή/και συσχετίσεις μεταξύ βιομορίων όπως οικογένειες πρωτεϊνών, κοινές δομές πρωτεϊνών κοινά μοτίβα ακολουθιών DNA και πρωτεϊνών. Εξειδικευμένες Β.Δ., κατηγορία στην οποία ανήκουν: Β.Δ. μικροσυστοιχιών που περιλαμβάνουν πληροφορίες για την έκφραση γονιδίων και πρωτεϊνών Β.Δ. Μεταβολικών μονοπατιών που περιέχουν πληροφορίες για τις χημικές αντιδράσεις που πραγματοποιούνται στο κύτταρο Βιβλιογραφικές βιολογικές βάσεις δεδομένων Βιολογικές βάσεις δεδομένων ιστοσελίδων που περιλαμβάνουν: Β.Δ. που περιλαμβάνουν ως εγγραφές βιολογικές βάσεις Συνδέσμους μεταξύ βιολογικών βάσεων δεδομένων.

33
Biological databases (source: wikipedia, http://en. wikipedia
Primary sequence databases The International Nucleotide Sequence Database (INSD) consists of the following databases. DDBJ (DNA Data Bank of Japan) EMBL Nucleotide DB (European Molecular Biology Laboratory) GenBank [1] (National Center for Biotechnology Information) These databanks represent the current knowledge about the sequences of all organisms. They interchange the stored information and are the source for many other databases. Meta-databases Strictly speaking a meta-database can be considered a database of databases, rather than any one integration project or technology. They collect data from different sources and usually makes them available in new and more convenient form, or with an emphasis on a particular disease or organism. Entrez[2] (National Center for Biotechnology Information) euGenes (Indiana University) GeneCards (Weizmann Inst.) SOURCE (Stanford University) mGen containing four of the world biggest databases GenBank, Refseq, EMBL and DDBJ - easy and simple program friendly gene extraction Bioinformatic Harvester[3] (Karlsruhe Institute of Technology) - Integrating 26 major protein/gene resources. MetaBase[4] (KOBIC) - A user contributed database of biological databases. ConsensusPathDB - A molecular functional interaction database, integrating information from 12 other databases. Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής
 consists of the following databases. DDBJ (DNA Data Bank of Japan) EMBL Nucleotide DB (European Molecular Biology Laboratory) GenBank [1] (National Center for Biotechnology Information) These databanks represent the current knowledge about the sequences of all organisms. They interchange the stored information and are the source for many other databases. Meta-databases. Strictly speaking a meta-database can be considered a database of databases, rather than any one integration project or technology. They collect data from different sources and usually makes them available in new and more convenient form, or with an emphasis on a particular disease or organism. Entrez[2] (National Center for Biotechnology Information) euGenes (Indiana University) GeneCards (Weizmann Inst.) SOURCE (Stanford University) mGen containing four of the world biggest databases GenBank, Refseq, EMBL and DDBJ - easy and simple program friendly gene extraction. Bioinformatic Harvester[3] (Karlsruhe Institute of Technology) - Integrating 26 major protein/gene resources. MetaBase[4] (KOBIC) - A user contributed database of biological databases. ConsensusPathDB - A molecular functional interaction database, integrating information from 12 other databases. Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής.")
34
Genome Databases These databases collect organism genome sequences, annotate and analyze them, and provide public access. Some add curation of experimental literature to improve computed annotations. These databases may hold many species genomes, or a single model organism genome. CAMERA Resource for microbial genomics and metagenomics Corn, the Maize Genetics and Genomics Database Ensembl provides automatic annotation databases for human, mouse, other vertebrate and eukaryote genomes. ERIC (Enteropathogen Resource Integration Center) Curated database containing annotated genome data for five enteropathogens - Escherichia coli, Shigella, Salmonella, Yersinia enterocolitica, and Y. pestis. Flybase, genome of the model organism Drosophila melanogaster MGI Mouse Genome (Jackson Lab.) JGI Genomes of the DOE-Joint Genome Institute provides databases of many eukaryote and microbial genomes. National Microbial Pathogen Data Resource. A manually curated database of annotated genome data for the pathogens Campylobacter, Chlamydia, Chlamydophila, Haemophilus, Listeria, Mycoplasma, Neisseria, Staphylococcus, Streptococcus, Treponema, Ureaplasma, and Vibrio. Saccharomyces Genome Database, genome of the yeast model organism. Viral Bioinformatics Resource Center Curated database containing annotated genome data for eleven virus families. The SEED platform for microbial genome analysis includes all complete microbial genomes, and most partial genomes. The platform is used to annotate microbial genomes using subsystems. Wormbase, genome of the model organism Caenorhabditis elegans Zebrafish Information Network, genome of this fish model organism. TAIR, The Arabidopsis Information Resource. UCSC_Malaria_Genome_Browser, genome of malaria causing species (Plasmodium_falciparumata and others) Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής
 Curated database containing annotated genome data for five enteropathogens - Escherichia coli, Shigella, Salmonella, Yersinia enterocolitica, and Y. pestis. Flybase, genome of the model organism Drosophila melanogaster. MGI Mouse Genome (Jackson Lab.) JGI Genomes of the DOE-Joint Genome Institute provides databases of many eukaryote and microbial genomes. National Microbial Pathogen Data Resource. A manually curated database of annotated genome data for the pathogens Campylobacter, Chlamydia, Chlamydophila, Haemophilus, Listeria, Mycoplasma, Neisseria, Staphylococcus, Streptococcus, Treponema, Ureaplasma, and Vibrio. Saccharomyces Genome Database, genome of the yeast model organism. Viral Bioinformatics Resource Center Curated database containing annotated genome data for eleven virus families. The SEED platform for microbial genome analysis includes all complete microbial genomes, and most partial genomes. The platform is used to annotate microbial genomes using subsystems. Wormbase, genome of the model organism Caenorhabditis elegans. Zebrafish Information Network, genome of this fish model organism. TAIR, The Arabidopsis Information Resource. UCSC_Malaria_Genome_Browser, genome of malaria causing species (Plasmodium_falciparumata and others) Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής.")
35
Genome Browsers Genome Browsers enable researchers to visualize and browse entire genomes (most have many complete genomes) with annotated data including gene prediction and structure, proteins, expression, regulation, variation, comparative analysis, etc. Annotated data is usually from multiple diverse sources. Integrated Microbial Genomes (IMG) system by the DOE-Joint Genome Institute UCSC Genome Bioinformatics Genome Browser and Tools (UCSC) Ensembl The Ensembl Genome Browser (Sanger Institute and EBI) GBrowse The GMOD GBrowse Project Pathway Tools Genome Browser X:Map A genome browser that shows Affymetrix Exon Microarray hit locations alongside the gene, transcript and exon data on a Google maps api Viral Genome Organizer (VGO) A genome browser providing visualization and analysis tools for annotated whole genomes from the eleven virus families in the VBRC (Viral Bioinformatics Resource Center) databases Apollo Genome Annotation Curation Tool A cross-platform, JAVA-based standalone genome viewer with enterprise-level functionality and customizations. The standard for many model organism databases. SEED viewer for visualizing and interrogating the SEED database of complete microbial genomes Integrated Genome Browser (IGB) A cross-platform, Java-based desktop genome viewer. Argo Genome Browser A fre and open source standalone Java-based genome browser for visualizing and manually annotating whole genomes. Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής
 with annotated data including gene prediction and structure, proteins, expression, regulation, variation, comparative analysis, etc. Annotated data is usually from multiple diverse sources. Integrated Microbial Genomes (IMG) system by the DOE-Joint Genome Institute. UCSC Genome Bioinformatics Genome Browser and Tools (UCSC) Ensembl The Ensembl Genome Browser (Sanger Institute and EBI) GBrowse The GMOD GBrowse Project. Pathway Tools Genome Browser. X:Map A genome browser that shows Affymetrix Exon Microarray hit locations alongside the gene, transcript and exon data on a Google maps api. Viral Genome Organizer (VGO) A genome browser providing visualization and analysis tools for annotated whole genomes from the eleven virus families in the VBRC (Viral Bioinformatics Resource Center) databases. Apollo Genome Annotation Curation Tool A cross-platform, JAVA-based standalone genome viewer with enterprise-level functionality and customizations. The standard for many model organism databases. SEED viewer for visualizing and interrogating the SEED database of complete microbial genomes. Integrated Genome Browser (IGB) A cross-platform, Java-based desktop genome viewer. Argo Genome Browser A fre and open source standalone Java-based genome browser for visualizing and manually annotating whole genomes. Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής.")
36
Protein Sequence Databases
UniProt[5] Universal Protein Resource (UniProt Consortium: EBI, Expasy, PIR) PIR Protein Information Resource (Georgetown University Medical Center (GUMC)) Swiss-Prot[6] Protein Knowledgebase (Swiss Institute of Bioinformatics) PEDANT Protein Extraction, Description and ANalysis Tool (Forschungszentrum f. Umwelt & Gesundheit) PROSITE Database of Protein Families and Domains DIP Database of Interacting Proteins (Univ. of California) Pfam Protein families database of alignments and HMMs (Sanger Institute) PRINTS PRINTS is a compendium of protein fingerprints (Manchester University) ProDom Comprehensive set of Protein Domain Families (INRA/CNRS) SignalP 3.0 Server for signal peptide prediction (including cleavage site prediction), based on artificial neural networks and HMMs SUPERFAMILY Library of HMMs representing superfamilies and database of (superfamily and family) annotations for all completely sequenced organisms Annotation Clearing House a project from the National Microbial Pathogen Data Resource Mathematical Model Databases CellML Biomodels Database [PCR / Real time PCR primer Databases PathoOligoDB: A free QPCR oligo database for pathogens Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής
 PIR Protein Information Resource (Georgetown University Medical Center (GUMC)) Swiss-Prot[6] Protein Knowledgebase (Swiss Institute of Bioinformatics) PEDANT Protein Extraction, Description and ANalysis Tool (Forschungszentrum f. Umwelt & Gesundheit) PROSITE Database of Protein Families and Domains. DIP Database of Interacting Proteins (Univ. of California) Pfam Protein families database of alignments and HMMs (Sanger Institute) PRINTS PRINTS is a compendium of protein fingerprints (Manchester University) ProDom Comprehensive set of Protein Domain Families (INRA/CNRS) SignalP 3.0 Server for signal peptide prediction (including cleavage site prediction), based on artificial neural networks and HMMs. SUPERFAMILY Library of HMMs representing superfamilies and database of (superfamily and family) annotations for all completely sequenced organisms. Annotation Clearing House a project from the National Microbial Pathogen Data Resource. Mathematical Model Databases. CellML. Biomodels Database. [PCR / Real time PCR primer. Databases. PathoOligoDB: A free QPCR oligo database for pathogens. Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής.")
37
Protein Structure Databases
Protein Data Bank[7] (PDB) (Research Collaboratory for Structural Bioinformatics (RCSB)) Protein Model Portal[8] (PMP) Meta database that combines several databases of protein structure models (Biozentrum, Basel, Switzerland) CATH Protein Structure Classification SCOP Structural Classification of Proteins SWISS-MODEL Server and Repository for Protein Structure Models ModBase Database of Comparative Protein Structure Models (Sali Lab, UCSF) Protein-proteiin Interactions BioGRID [9] A General Repository for Interaction Datasets (Samuel Lunenfeld Research Institute) STRING: STRING is a database of known and predicted protein-protein interactions. (EMBL) DIP Database of Interacting Proteins BIND Biomolecular Interaction Network Database Metabolic pathway Databases BioCyc Database Collection including EcoCyc and MetaCyc KEGG PATHWAY Database[10] (Univ. of Kyoto) MANET database [11] (University of Illinois) Reactome[12] (Cold Spring Harbor Laboratory, EBI, Gene Ontology Consortium) Microarray Databases ArrayExpress (European Bioinformatics Institute) Gene Expression Omnibus (National Center for Biotechnology Information) maxd (Univ. of Manchester) Stanford Microarray Database (SMD) (Stanford University) GPX(Scottish Centre for Genomic Technology and Informatics) Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής
 (Research Collaboratory for Structural Bioinformatics (RCSB)) Protein Model Portal[8] (PMP) Meta database that combines several databases of protein structure models (Biozentrum, Basel, Switzerland) CATH Protein Structure Classification. SCOP Structural Classification of Proteins. SWISS-MODEL Server and Repository for Protein Structure Models. ModBase Database of Comparative Protein Structure Models (Sali Lab, UCSF) Protein-proteiin Interactions. BioGRID [9] A General Repository for Interaction Datasets (Samuel Lunenfeld Research Institute) STRING: STRING is a database of known and predicted protein-protein interactions. (EMBL) DIP Database of Interacting Proteins. BIND Biomolecular Interaction Network Database. Metabolic pathway Databases. BioCyc Database Collection including EcoCyc and MetaCyc. KEGG PATHWAY Database[10] (Univ. of Kyoto) MANET database [11] (University of Illinois) Reactome[12] (Cold Spring Harbor Laboratory, EBI, Gene Ontology Consortium) Microarray Databases. ArrayExpress (European Bioinformatics Institute) Gene Expression Omnibus (National Center for Biotechnology Information) maxd (Univ. of Manchester) Stanford Microarray Database (SMD) (Stanford University) GPX(Scottish Centre for Genomic Technology and Informatics) Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής.")
38
Specialized databases
Genome Database for Rosaceae (International Genomics and Genetics Database for Rosaceous crops) XTractorDiscovering Newer Scientific Relations Across PubMed Abstracts. A tool to obtain manually annotated relationships for Proteins, Diseases, Drugs and Biological Processes as they get published in PubMed. BIOMOVIE (ETH Zurich) movies related to biology and biotechnology CGAP Cancer Genes (National Cancer Institute) Clone Registry Clone Collections (National Center for Biotechnology Information) DBGET H.sapiens (Univ. of Kyoto) GDB Hum. Genome Db (Human Genome Organisation) SHMPD The Singapore Human Mutation and Polymorphism Database NCBI-UniGene (National Center for Biotechnology Information) OMIM Inherited Diseases (Online Mendelian Inheritance in Man) Off. Hum. Genome Db (HUGO Gene Nomenclature Committee) HGMD disease-causing mutations (HGMD Human Gene Mutation Database) PhenCode linking human mutations with phenotype List with SNP-Databases p53 The p53 Knowledgebase Edinburgh Mouse Atlas HvrBase++ Human and primate mitochondrial DNA PolygenicPathways Genes and risk factors implicated in Alzheimer's disease, Bipolar disorder or Schizophrenia Connectivity map Transcriptional expression data and correlation tools for drugs CTD The Comparative Toxicogenomics Database describes chemical-gene-disease interactions DiProDB A database to collect and analyse thermodynamic, structural and other dinucleotide properties. GyDB The Gypsy Database of Mobile Genetic Elements (Universitat de València) INTERFEROME The Database of Interferon Regulated Genes Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής
 XTractorDiscovering Newer Scientific Relations Across PubMed Abstracts. A tool to obtain manually annotated relationships for Proteins, Diseases, Drugs and Biological Processes as they get published in PubMed. BIOMOVIE (ETH Zurich) movies related to biology and biotechnology. CGAP Cancer Genes (National Cancer Institute) Clone Registry Clone Collections (National Center for Biotechnology Information) DBGET H.sapiens (Univ. of Kyoto) GDB Hum. Genome Db (Human Genome Organisation) SHMPD The Singapore Human Mutation and Polymorphism Database. NCBI-UniGene (National Center for Biotechnology Information) OMIM Inherited Diseases (Online Mendelian Inheritance in Man) Off. Hum. Genome Db (HUGO Gene Nomenclature Committee) HGMD disease-causing mutations (HGMD Human Gene Mutation Database) PhenCode linking human mutations with phenotype. List with SNP-Databases. p53 The p53 Knowledgebase. Edinburgh Mouse Atlas. HvrBase++ Human and primate mitochondrial DNA. PolygenicPathways Genes and risk factors implicated in Alzheimer s disease, Bipolar disorder or Schizophrenia. Connectivity map Transcriptional expression data and correlation tools for drugs. CTD The Comparative Toxicogenomics Database describes chemical-gene-disease interactions. DiProDB A database to collect and analyse thermodynamic, structural and other dinucleotide properties. GyDB The Gypsy Database of Mobile Genetic Elements (Universitat de València) INTERFEROME The Database of Interferon Regulated Genes. Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής.")
39
Wiki style databases www.EcoliHub.org/EcoliWiki Gene Wiki OpenWetWare
PDBWiki Proteopedia Topsan WikiGenes WikiPathways YTPdb CHDwiki WikiProfessional Εισαγωγή στη Βιοπληροφορική Κατερίνα Περδικούρη, Χρήστος Μακρής

40
Βάσεις Βιολογικών Δεδομένων (1)
GenBank: NCBI ( GenBank® is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences. There are approximately 37,893,844,733 bases in 32,549,400 sequence records as of February Δείγμα εγγραφής (National Center of Biotechnology Information) PIR: Protein Information Resource ( PIR produces the Protein Sequence Database (PSD) of functionally annotated protein sequences, which grew out of the Atlas of Protein Sequence and Structure ( ) edited by Margaret Dayhoff and has been incorporated into an integrated knowledge base system of value-added databases and analytical tools. Swiss-Prot + TrEMBL: Swiss-Prot.htm ( Swiss-Prot is a curated protein sequence database which strives to provide a high level of annotation (such as the description of the function of a protein, its domains structure, post-translational modifications, variants, etc.), a minimal level of redundancy and high level of integration with other databases TrEMBL is a computer-annotated supplement of Swiss-Prot that contains all the translations of EMBL nucleotide sequence entries not yet integrated in Swiss-Prot.
 PIR: Protein Information Resource ( PIR produces the Protein Sequence Database (PSD) of functionally annotated protein sequences, which grew out of the Atlas of Protein Sequence and Structure ( ) edited by Margaret Dayhoff and has been incorporated into an integrated knowledge base system of value-added databases and analytical tools. Swiss-Prot + TrEMBL: Swiss-Prot.htm ( Swiss-Prot is a curated protein sequence database which strives to provide a high level of annotation (such as the description of the function of a protein, its domains structure, post-translational modifications, variants, etc.), a minimal level of redundancy and high level of integration with other databases. TrEMBL is a computer-annotated supplement of Swiss-Prot that contains all the translations of EMBL nucleotide sequence entries not yet integrated in Swiss-Prot.")
41
Βάσεις Βιολογικών Δεδομένων (2)
PRODOM: ( ProDom is a comprehensive set of protein domain families automatically generated from the SWISS-PROT and TrEMBL sequence databases PROTOMAP: ( An exhaustive classification of all the proteins in the SWISSPROT and TrEMBL databases, into groups of related proteins. The analysis uses transitivity to identify homologous proteins, and within each group, every two members are either directly or transitively related. PRINTS: ( PRINTS is a compendium of protein fingerprints. A fingerprint is a group of conserved motifs used to characterise a protein family; its diagnostic power is refined by iterative scanning of a SWISS-PROT/TrEMBL composite.

42
Βάσεις Βιολογικών Δεδομένων (3)
PROSITE: Prosite ( PROSITE is a database of protein families and domains. It consists of biologically significant sites, patterns and profiles that help to reliably identify to which known protein family (if any) a new sequence belongs. PDB-Protein Data Bank: PDB ( The PDB is the single worldwide repository for the processing and distribution of 3-D structure data of large molecules of proteins and nucleic acids. GDB: Genome Mapping Data ( Genome Mapping Data from the Human Genome Initiative
 a new sequence belongs. PDB-Protein Data Bank: PDB ( The PDB is the single worldwide repository for the processing and distribution of 3-D structure data of large molecules of proteins and nucleic acids. GDB: Genome Mapping Data ( Genome Mapping Data from the Human Genome Initiative.")
43
Βάσεις Βιολογικών Δεδομένων (4)
TIGR: The Institute for Genomic Research (TIGR) is a not-for-profit research institute whose primary research interests are in structural, functional and comparative analysis of genomes and gene products from a wide variety of organisms including viruses, eubacteria (both pathogens and non-pathogens, archaea (the so-called third domain of life), and eukaryotes (plants, animals, fungi and protists such as the malarial parasite). SCOP: ( Structural Classification of Proteins
 is a not-for-profit research institute whose primary research interests are in structural, functional and comparative analysis of genomes and gene products from a wide variety of organisms including viruses, eubacteria (both pathogens and non-pathogens, archaea (the so-called third domain of life), and eukaryotes (plants, animals, fungi and protists such as the malarial parasite). SCOP: ( Structural Classification of Proteins.")
44
Ένα παράδειγμα: PDB data entry example

45
Sequence Database Searching
Βήματα καθορισμού πρωτεϊνικής ακολουθίας Σύγκριση της νέας ακολουθίας με PROSITE και BLOCKS για εύρεση well-characterized sequence motifs. Ψάξιμο στις DNA και protein sequence databases (Genbank, Swiss-Prot, etc.) για εντοπισμό ακολουθιών τοπικά παρόμοιων (με χρήση ενός κριτηρίου τοπικής ομοιότητας) – χρήση FASTA και BLAST Εάν τα παραπάνω ψαξίματα δίνουν ενδιαφέρον αποτέλεσμα, τότε καταφεύγουμε στη χρήση της τεχνικής του δυναμικού προγραμματισμού. Όταν χρειαστεί να εμπλακούν amino acid substitution matrices, συνήθως χρησιμοποιείται μία παραλλαγή του Dayhoff PAM matrix και του BLOSUM matrix.
 για εντοπισμό ακολουθιών τοπικά παρόμοιων (με χρήση ενός κριτηρίου τοπικής ομοιότητας) – χρήση FASTA και BLAST. Εάν τα παραπάνω ψαξίματα δίνουν ενδιαφέρον αποτέλεσμα, τότε καταφεύγουμε στη χρήση της τεχνικής του δυναμικού προγραμματισμού. Όταν χρειαστεί να εμπλακούν amino acid substitution matrices, συνήθως χρησιμοποιείται μία παραλλαγή του Dayhoff PAM matrix και του BLOSUM matrix.")
46
Ο Αλγόριθμος BLAST (Συντηρείται από το NCBI) Αλγόριθμος Είδος query
BLAST: Basic Local Alignment Search Tool, Altschul et. Al. 1990 Βασική ιδέα: εντοπισμός κοινών υπο-ακολουθιών ίδιου μήκους (segment pairs) που εμφανίζονται και στη δοσμένη ακολουθία μικρού μήκους (input query sequence) και στο σύνολο των ακολουθιών μίας βάσης δεδομένων. Στη συνέχεια επέκταση για εύρεση maximal segment pairs Αλγόριθμος Είδος query Είδος sequence BLASTP Πρωτείνη Πρωτεϊνη BLASTN Νουκλεοτίδιο BLASTX TBLASTN TBLASTX
 που εμφανίζονται και στη δοσμένη ακολουθία μικρού μήκους (input query sequence) και στο σύνολο των ακολουθιών μίας βάσης δεδομένων. Στη συνέχεια επέκταση για εύρεση maximal segment pairs. Αλγόριθμος. Είδος query. Είδος sequence. BLASTP. Πρωτείνη. Πρωτεϊνη. BLASTN. Νουκλεοτίδιο. BLASTX. TBLASTN. TBLASTX.")
48
O Αλγόριθμος FASTA (φιλοξενείται στο ΕΒΙ)
FASTΑ: Fast – All, Lipman et al. 1985 Κεντρική ιδέα: εύρεση μικρών λέξεων (words ή k-tuples) που εμφανίζονται και στις δύο ακολουθίες. Στην περίπτωση πρωτεϊνικών ακολουθιών το μήκος των λέξεων είναι 1-2 βάσεις ενώ για ακολουθίες DNA το μήκος μίας λέξης μπορεί να φτάνει στις 6 βάσεις
 που εμφανίζονται και στις δύο ακολουθίες. Στην περίπτωση πρωτεϊνικών ακολουθιών το μήκος των λέξεων είναι 1-2 βάσεις ενώ για ακολουθίες DNA το μήκος μίας λέξης μπορεί να φτάνει στις 6 βάσεις.")
49
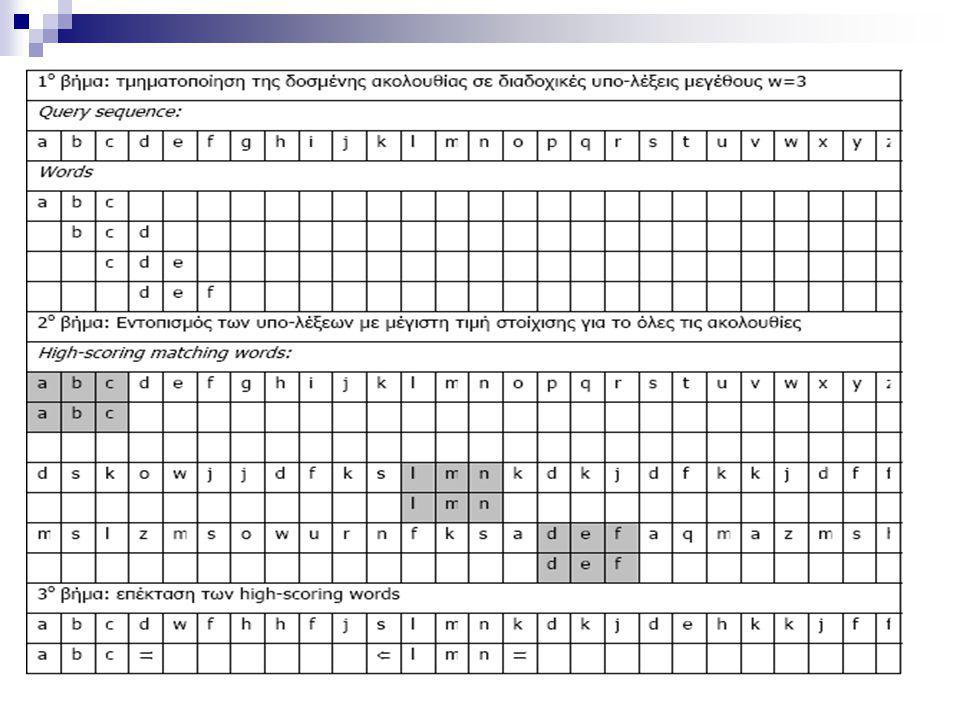
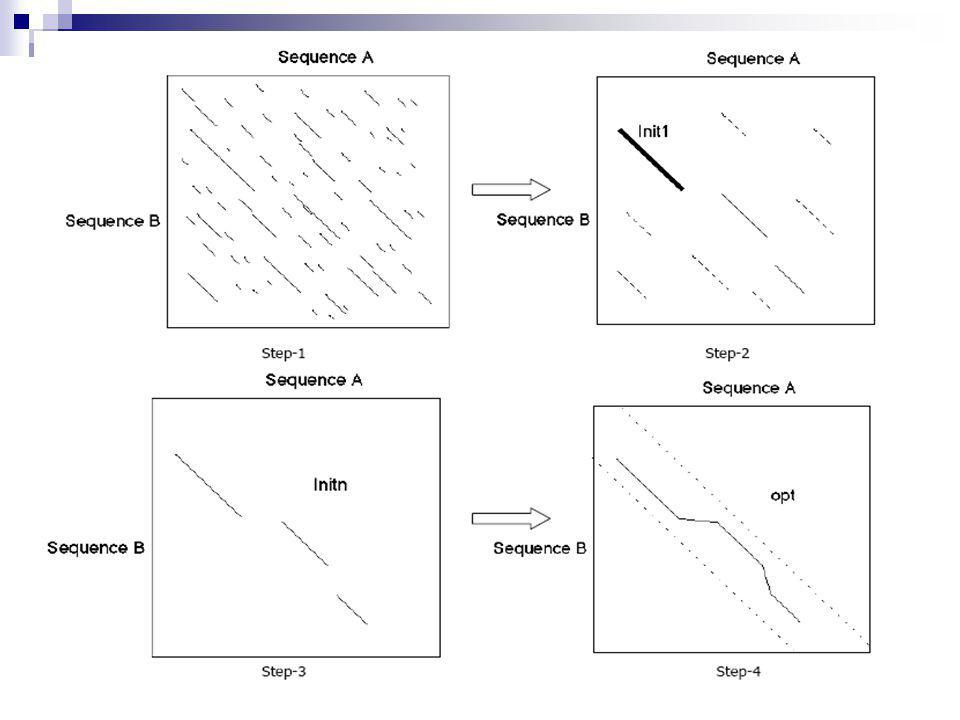
Τα βήματα του αλγορίθμου FASTA
1o βήμα: αναζητούμε λέξεις μήκους ktup στον πίνακα δυναμικού προγραμματισμού: ‘hot spots’ (pairs (i,j) ) 2ο βήμα: εντοπίζουμε τις δέκα καλύτερες διαγώνιες τροχιές – diagonal runs από ‘hot-spots’ στον πίνακα (a hot spot define the (i-j)-diagonal, the score is the sum of scores of hot-spots plus weighted decreasing as the distance increases) 3ο βήμα: συνδυάζουμε «καλές υπο-στοιχίσεις» 4ο βήμα: παράγουμε το βέλτιστο μονοπάτι
 ) 2ο βήμα: εντοπίζουμε τις δέκα καλύτερες διαγώνιες τροχιές – diagonal runs από ‘hot-spots’ στον πίνακα. (a hot spot define the (i-j)-diagonal, the score is the sum of scores of hot-spots plus weighted decreasing as the distance increases) 3ο βήμα: συνδυάζουμε «καλές υπο-στοιχίσεις» 4ο βήμα: παράγουμε το βέλτιστο μονοπάτι.")
51
Άλλα Θέματα Amino Acid Substitution Matrices
Dayhoff PAM (Point Accepted Mutation) BLOSUM (derived from the BLOCKS database) BLOCKS is a database of protein motifs that attempts to represent the most highly conserved substrings in aminoacid sequences or related preoteins.
 BLOSUM (derived from the BLOCKS database) BLOCKS is a database of protein motifs that attempts to represent the most highly conserved substrings in aminoacid sequences or related preoteins.")
52
Indexing Approaches k-gram indexing direct indexing
vector space indexing

53
k-gram Indexing Hash tables (FASTP, FASTA, BLAST, BL2SEQ, PSI-BLAST, MegaBLAST, BLASTZ, WU-BLAST, BLAT, SSAHA, SENSEI) ed-tree (the gram size k, the skip interval Δ (which start positions matter), the segment length vector H=[h1, …, ht], where sumi(hi)=k) -- For each sequence, the algorithm generates all k-grams with Δ skips -- Each gram is partitioned according to H, and the partitions are inserted into the ed-tree. The search algorithm partitions accordingly the query string, and initiates a search procedure, beginning from the root. Some important properties: (1) the k-grams are usually longer than that of BLAST, (2) they allow inexact k-gram matches, (3) only one k-gram out of Δ k-grams is indexed, Ed-tree is used by CHAOS, LAGAN, DIALIGN.
, the segment length vector H=[h1, …, ht], where sumi(hi)=k) -- For each sequence, the algorithm generates all k-grams with Δ skips. -- Each gram is partitioned according to H, and the partitions are inserted into the ed-tree. The search algorithm partitions accordingly the query string, and initiates a search procedure, beginning from the root. Some important properties: (1) the k-grams are usually longer than that of BLAST, (2) they allow inexact k-gram matches, (3) only one k-gram out of Δ k-grams is indexed, Ed-tree is used by CHAOS, LAGAN, DIALIGN.")
54
Direct Indexing Suffix trees -- MUMmer, AVID, REPuter, MGA, QUASAR
VP-trees (Vantage Point tree)
")
55
MUMmer Detecting MUM (Maximal Unique Matches), that is pair of substrings (x’,y’) that exactly match and there is no other matching pair that contains x’, y’ simultaneously (just use the GST(x,y)) Find the backbone of the alignment: all the pairs (x’,y’) are sorted in increasing order of the position of x’. Next the longest sequence of MUMs whose subsequences from x, y are in sorted order is found. These form the backbone Closing gaps. The gaps between consecutive MUMs are aligned with the help of Smith-Waterman Similar tools are AVID, REPuter, MGA.
, that is pair of substrings (x’,y’) that exactly match and there is no other matching pair that contains x’, y’ simultaneously (just use the GST(x,y)) Find the backbone of the alignment: all the pairs (x’,y’) are sorted in increasing order of the position of x’. Next the longest sequence of MUMs whose subsequences from x, y are in sorted order is found. These form the backbone. Closing gaps. The gaps between consecutive MUMs are aligned with the help of Smith-Waterman. Similar tools are AVID, REPuter, MGA.")
56
VP-trees (Vantage Point tree)
The VP-tree has been adapted to sequence databases where the distance is the edit distance or the block edit distance. It can be applied to other distance functions as long as they are almost metric. The algorithm takes a database, D={s1,…,sn}, and chooses a sequence s as the root, while the median of the distances to s is computed. The two sets are: (i) the sequences that are closer to s than the median, (ii) the rest of the sequences. Given a query q, the sequences at distance r are found as follows: First, q is compared to the vantage sequence s, at the root node. Let M be the median distance. 1. If d(q,s)≤r, s is inserted to the result set 2. If d(q,s)≤r+M, then the left child is searched recursively. 3. If d(q,s)≥M-r, then the right child is searched recursively.
 the sequences that are closer to s than the median, (ii) the rest of the sequences. Given a query q, the sequences at distance r are found as follows: First, q is compared to the vantage sequence s, at the root node. Let M be the median distance. 1. If d(q,s)≤r, s is inserted to the result set. 2. If d(q,s)≤r+M, then the left child is searched recursively. 3. If d(q,s)≥M-r, then the right child is searched recursively.")
57
Vector Space Indexing These index structures, map sequences or subsequences to vectors in a vector space. There exist two important index structures: SST (Sequence Search Tree) MRS (Multi Resolution String) index
 MRS (Multi Resolution String) index.")
58
SST Vector Space Mapping (window size w, shift amount Δ, tuple size k)
The parameter k, determines the size of the computed vector (for alphabet size σ this vector has size σκ ) The produced vectors are stored in a so called centroid structure. A query is performed as follows: a query sequence is first divided into subsequences of window size w, using a shift amount of Δ=w/2. Each of the produced query vectors is then searched on the index structure starting from the root node.
 The produced vectors are stored in a so called centroid structure. A query is performed as follows: a query sequence is first divided into subsequences of window size w, using a shift amount of Δ=w/2. Each of the produced query vectors is then searched on the index structure starting from the root node.")
Παρόμοιες παρουσιάσεις












 polynomial (X, X). polynomial (Term, X) :- number (Term).>")






 «ΑΓΙΟΣ ΑΘΑΝΑΣΙΟΣ.>")