Κατέβασμα παρουσίασης
1
Λοιμώδη νοσήματα ή λοιμώξεις (Infectious diseases)
Προκαλούνται από διείσδυση, εγκατάσταση και πολλαπλασιασμό μέσα στο σώμα του ανθρώπου παθογόνων μικροοργανισμών Οι εκδηλώσεις τους οφείλονται στην τοπική ή συστηματική αντίδραση του οργανισμού έναντι του παθογόνου παράγοντα 1

4
Κατάταξη παθογόνων οργανισμών
4

6
6

7
Μικροβιακό κύτταρα (τυπικό προακρυωτικό κύτταρο)
7

8
8

11
ΟΡΙΣΜΟΙ Αντιμικροβιακά φάρμακα Αντιβιοτικά Χημειοθεραπευτικά
Από μκροοργανισμούς ή συνθετικώς (εν μέρει ή στο σύνολο) παραγόμενα φάρμακα τα οποία καταστρέφουν ή εμποδίζουν τον πολλαπλασιασμό άλλων μικροοργανισμών Αντιβιοτικά παράγονται από μικρόβια ή μύκητες Χημειοθεραπευτικά συνθετικώς παραγόμενα Αντιικά φάρμακα Αντιμυκητιασικά φάρμακα 11
 παραγόμενα φάρμακα τα οποία καταστρέφουν ή εμποδίζουν τον πολλαπλασιασμό άλλων μικροοργανισμών. Αντιβιοτικά. παράγονται από μικρόβια ή μύκητες. Χημειοθεραπευτικά. συνθετικώς παραγόμενα. Αντιικά φάρμακα. Αντιμυκητιασικά φάρμακα. 11.")
13
Η ΔΡΑΣΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΟ ΜΙΚΡΟΒΙΑΚΟ ΚΥΤΤΑΡΟ

14
Η ΔΡΑΣΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΑ ΡΙΒΟΣΩΜΑΤΑ ΤΟΥ ΒΑΚΤΗΡΙΑΚΟΥ ΚΥΤΤΑΡΟΥ

15
ΙΣΤΟΡΙΑ ΑΝΤΙΒΙΟΤΙΚΩΝ 1910: Salvarsan (παράγωγο αρσενικού) από Paul Ehlrich 1928: Penicillin (παράγωγο μύκητα) από Alexander Fleming 1932: σουλφοναμίδες (prontosil) από Gerhard Domagk 1941: Καθαρή μορφή πενικιλλίνης από Ernst Chain και Howard Florey Χρήση πενικιλίνης κατά το 2ο παγκόσμιο πόλεμο σήμερα: Παραγωγή μεγάλου αριθμού αντιβιοτικών.
 από Gerhard Domagk. 1941: Καθαρή μορφή πενικιλλίνης από Ernst Chain και Howard Florey. Χρήση πενικιλίνης κατά το 2ο παγκόσμιο πόλεμο σήμερα: Παραγωγή μεγάλου αριθμού αντιβιοτικών.")
16
ΟΙ ΕΦΕΥΡΕΤΕΣ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ
Fleming receiving the Nobel prize from King Gustav V of Sweden (right), 1945 Regional Research Laboratory in Peoria, Illinois became the first site for commercial production of penicillin [5]. By mid-1944, when the Allies invaded France, large supplies of the yellow liquid were available. When treated with penicillin, 95% of the wounded lived. The Nobel Prize in Medicine was awarded to Fleming, Florey, and Chain in 1945. Selman A. Waksman, a Russian immigrant to the United States, gave the name ‘‘antibiotics’’ to chemicals (produced by soil-borne fungi and microorganisms) that destroy or slow the growth of other microbes. Waksman spent his lifetime hunting for ‘‘antibiotic’’-producing micro-organisms and in 1943 found a mold that was able to kill Tubercle bacilli. He called this aminoglycoside streptomycin. On November 20, 1944, streptomycin was administered to a young woman who had advanced pulmonary tuberculosis at the Mayo Clinic in Rochester, Minnesota. Her life was saved by streptomycin. Dr. Waksman received the Nobel Prize in 1952 for its discovery. The rest is history: awakening to the possibilities and confronting the challenges Subsequently, more broad-spectrum penicillins and other aminoglycosides were developed, followed by more antibiotic classes. More than 5000 antibiotics are now known. Approximately a thousand of these have been carefully investigated, and about 100 are currently used to treat infections. Most are produced by actinomycetes and bacteria, many of which are then chemically modified (semisynthetic). Others are completely synthetic. The b-lactams Penicillin, the prototype b-lactam, is a 6-aminopenicillanic acid consisting of a thiazolidine ring, an attached b-lactam ring, and a side chain [6]. Manipulations of the side chain have altered b-lactamase susceptibility, antibacterial spectrum, and pharmacokinetic properties. Other groups of antibacterial agents that contain the b-lactam ring include cephalosporins, carbapenems, and monobactams. In actively dividing bacteria, the b-lactams inhibit enzymes (transpeptidase, carboxypeptidase, and endopeptidase) located beneath the cell wall that are termed the ‘‘penicillin-binding proteins.’’ This inhibition prevents the development of normal peptidoglycan structure, because these enzymes are involved in creating the cross-linkage between the peptide chains. Various bacteria differ in the permeability of their cell walls to antibiotics and the type and concentration of penicillinbinding proteins. Subsequent activation of the endogenous autolytic system of bacteria by b-lactams initiates cell lysis and death [7]. Penicillins The penicillin family of antibiotics remains an important part of today’s antimicrobial armamentarium. Penicillins are bactericidal against most ANTIBIOTICS: PAST, PRESENT, FUTURE 1051 It was in the mid-nineteenth century that Louis Pasteur observed that some micro-organisms destroy othersdthe phenomenon that later came to be known as antibiosis or ‘‘against life.’’ The search for antimicrobial chemical agents revealed that antiseptics were too toxic for anything but surface use on wounds. German bacteriologist Paul Ehrlich systematically tested chemical agents, searching for the ‘‘magic bullet’’ that could be taken internally, but he ended up with only a high-risk arsenic-based treatment for syphilis. Alexander Fleming in London had been looking for antibacterial agents in human secretions. The discovery of the antibacterial activity of the enzyme lysozyme was made because of an accidental sneeze on a petri dish [3]. Fleming observed that, when bacteria later formed colonies on the plate, none developed in the spots occupied by mucus. Further tests showed that lysozyme acted mostly against harmless organisms. In 1928, serendipity made another notable visit to Fleming’s laboratory at St. Mary’s Hospital in London. He had left a culture plate of Staphylococci uncovered in his laboratory while on a vacation. On his return, he noticed mold in the petri dish along with a clear space between the Staphylococci and the bluegreen spotted mold. It was the classic example of what Pasteur had referred to as fortune’s accommodating a willing mind. Fleming identified the mold as Penicillium natatum, a culture filtrate of which was able to kill bacteria. He named the agent in the filtrate penicillin. Because of a paucity of financial resources and Fleming’s modest ambitions (according to some historians), it took another 12 years for penicillin to emerge as the greatest medical advance of the twentieth (or any other) century. But the golden age of anti-infective medicine actually began in 1934. Gerhard Domagk, a German pharmacologist, discovered that a dye used to tint cloth cured streptococcal infections in mice. His own dying daughter survived a streptococcal infection after he injected her with the dye. Daniel Bovert, a Swiss-born scientist, identified the active compound as sulfanilamide. Domagk was awarded the Nobel Prize in Medicine in 1939. Florey and Chain, working at Oxford University, were interested to note that staphylococci, though resistant to sulfanamides and lysozyme, were apparently sensitive to the penicillium mold [4]. World War II provided a crucial spur to and much-needed resources for research on antimicrobial agents. Staphylococcal infections and gas gangrene were killing more men than the immediate organ damage caused by shell and bullet wounds. In the spring of 1940, Florey and Chain were able to make a small amount of yellowish-brown powder from Fleming’s mold. This first sample of ‘‘penicillin powder’’ was a million times more potent than Fleming’s original filtrate. In 1941, the Fermentation Division of the newly created Northern Regional Research Laboratory in Peoria, Illinois became the first site for Dr. Waksman received the Nobel Prize in 1952 for its discovery.Antibiotics are compounds that act to kill or inhibit the growth of bacteria1. The etymology of the term can be broken down into two roots: the prefix “anti-” meaning “opposed to” or “preventing” and “biotic” coming from the Greek word for life. In nature, various microbes and fungi secrete these compounds to gain an advantage in their microenvironment and it is from these very organisms that antibiotics are commonly use isolated1. The Discovery of Antibiotics The stories of the discovery of antibiotics are dramatic and full of human interest, both on a global and personal scale. Two brief recounts are given below (please see reference 2 for an excellent overview of antibiotics). Alexander Fleming is popularly thought to have been the discoverer of penicillin. He is certainly the first researcher to have recognized its potential. In 1928 Fleming discovered that a blue mold (Penicillium) was able to lyse bacterial Staphylococci cells. Fleming determined that Penicillium produced some compound that caused the bacterial cells to lyse. He called this compound penicillin. And how he came to these conclusions is a now famous story of fortuitous chance3. Fleming had returned to the lab after a holiday to discover that some culture plates of Staphylococci had become contaminated. It was an accidental growth of Penicillium, but luckily one he did not throw away. His further observation that the contaminating mold was able to kill Staphylococci led to his being awarded the Nobel Prize for Medicine in Howard Florey and Ernest Chain were also honoured with the Nobel Prize for developing a way to produce large quantities of penicillin. Gerhard Domagk discovered the first sulfa drugs in The pharmaceutical company Bayer had hired him to work on the problem of infectious diseases caused by the bacterium Streptococcus pyogenes. In various trials designed to determine the effectiveness of various compounds for bacterial killing, Domagk discovered that a dye called prontosil rubrum prevented S. pyogenes infection in mice. In 1935 Domagk’s daughter was gravely ill due to S. pyogenes infection. The infection was advancing so aggressively that doctors were considering amputating her arm, but instead she was treated with prontosil rubrum (well before complete clinical trials of the drug had been completed). She made a full recovery. Domagk received the Nobel Prize in Mechanism of Action Being such an important medical compound the mechanisms by which antibiotics kill bacteria have been under scrutiny for decades, with such studies being instrumental in the design of new and improved compounds. There are three general modes of antibiotic activity: (1) interference with the cell wall, (2) interference with nucleic acid synthesis, and (3) interference with protein synthesis Figure 1. A basic schematic showing the arrangement of the cell wall in relation to the plasma membrane of a bacterial cell. The thickness and composition of the cell wall is different between the Gram-positive and Gram-negative cells. Interference with the Cell Wall There is a multitude of ways to classify bacteria, but one of the more common methods is as either Gram-positive or Gram-negative cells4. These Gram designations are based on a differential staining assay, with the bacteria that stain dark being referred to as Gram-positive, while those that do not stain dark being referred to as Gram-negative. This difference in stain intensity and thus designation does have a physical basis, which is linked to the cell membrane. In every bacterial cell the plasma membrane encases the contents of the cell (referred to as the cytoplasm) and directly outside the plasma membrane is an additional exterior cell wall (See Figure 1). The plasma membrane of the cell is pressed tightly against the cell wall due to turgor pressure. It is the cell wall that determines the shape of the cells, the strength of which is provided by peptidoglycan5 (the major structural component of the cell wall). Gram-positive bacteria have thick cell walls composed primarily of this substance; where as Gram-negative bacteria have multi-layer cell walls that are thinner than those of the Gram-positive cells. Peptidoglycan is a lattice-like macromolecule composed of repeated sugar units that are cross-linked together, with the “glycans” of peptidoglycan forming polysaccharide strands that run parallel to each other (See Figure 2). These polysaccharide strands are composed of alternating units of two sugars: N-acetylmuramic acid (commonly referred to as NAM) and N-acetylglucosamine (commonly referred to as NAG). Additionally, peptidoglycan contains two peptides that cross-link these long polysaccharide strands and are composed of alanine, glutamic acid, lysine and alanine. Tetra-peptides linking the polysaccharide strands are themselves linked by a penta-peptide of five glycine residues that runs from the lysine residue of one tetrapeptide to the terminal alanine residue of another tetra-peptide. Figure 2 shows how these three different components (the polysaccharide strands, the tetra-peptide and the penta-peptide) come together to form peptidoglycan and give this macromoleclue its strength Figure 2. A schematic of peptidoglycan’s structure. The NAM and NAG sugars are shown as green and blue spheres respectively. The tetrapeptides linked to NAM are cross-linked by a pentaglycine peptide, shown as red lines linking the D-glutamine (L) to the D-alanine (A).] Peptidoglycan is synthesized in three stages in three different parts of the cell. Firstly, the NAM sugar is linked to the alanine, glutamic acid, lysine and alanine precursor in the cytoplasm, forming the basic subunit of peptidoglycan. Secondly, the NAM/peptide is linked to the NAG sugar at the cell membrane. And lastly, now at the cell wall, the newly synthesized peptidoglycan subunit is transferred to the growing point of the cell wall’s peptidoglycan by a bond between the old peptidoglycan and the new NAM-NAG disaccharide. Here the lateral cross-linking by the pentaglycine peptide can occur. Antibiotics are active at every step of peptidoglycan synthesis. Among the most famous of the antibiotics to interfere with peptidoglycan synthesis are the b-lactams, of which penicillin is an example6. Penicillins resemble the terminal amino acids of the NAM+tetrapeptide precursor synthesized in the cytoplasm and actually bind the enzyme that catalyzes the pentaglycine cross-linking reaction. This binding prevents the cross-linking reaction from occurring and thus weakens the cell wall. Eventually, the turgor pressure of the cell causes the cell to lose its shape and eventually burst if the surrounding solution is hypotonic. Go to to see a short movie showing the activity of penicillin on Escherichia coli. On the flip side of this coin, bacteria can become resistant to penicillin by three strategies: the hydrolysis of penicillin, the acquisition of proteins with a reduced affinity for penicillin or the reduced uptake of penicillin. The hydrolysis of penicillin is catalyzed by enzymes called b-lactamases, which ring structure of penicillin and prevent it from mimicing the structure of the peptidoglycan precursor. Interference with Nucleic Acid Synthesis Antibiotics are frequently active against nucleic acid synthesis within the bacterial cell1. This includes inhibition or interference of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis. The antibiotics that act in this manner are frequently analogs of essential metabolites of the cell, compounds that block DNA template activity during synthesis of new DNA, or compounds that block the transcription of DNA into RNA An example is the sulfonamides, commonly known as sulfa drugs, which are derivatives of dyes and resemble the compound folate. Folate is a coenzyme (a substance required for the proper functioning of enzymes) that is essential for cell growth and in bacterial cells is a precursor for the synthesis of amino acids and nucleic acids. Most bacteria synthesize folate from scratch, whereas mammalian cells cannot do this and must transport folate, which has been made by other sources, into their cells. This metabolic difference between bacterial cells and mammalian cells makes folate biosynthesis a convenient target for antibiotics, as this pathway is specific to bacterial cells. The sulfa drugs mimic one of the folate precursors, competing with the precursor for the enzyme involved in the next step of folate synthesis. If the normal folate precursor cannot bind the enzyme (because the sulfa drug is there), folate synthesis is blocked. Thus, the bacteria cannot synthesize the nucleic acids and some of the amino acids necessary for cell survival and perish. The method by which an antibiotic affects nucleic acid synthesis is actually quite diverse. For example, the coiling of the bacterial chromosome can be attacked. It is quite a long structure and its great size requires that this it is efficiently packed into the cell by supercoiling. The tightly coiled DNA must be uncoiled and relaxed in order for the DNA or RNA polymerases to gain access to the DNA template, a process accomplished by enzymes known as topoisomerases. All three types of enzymes, the RNA and DNA polymerases and topoisomerases, are antibiotic targets. Drugs called quinolones target a topoisomerase known as ‘DNA gyrase’, which normally uncoils DNA by cutting the two strands and then passing a section of the double helix through the gap. Quinolones bind to the cut strands of DNA, preventing the re-annealing (gluing back together) of the cut DNA strands with the parent strands. An alternative is the nitroimidazoles, drugs that cleave the double-stranded DNA template by producing radical ions. Cleavage of the DNA template interferes with both DNA replication and RNA synthesis. Classes of drugs known as rifamycins actually specifically inhibit RNA synthesis by binding to the RNA polymerase and preventing the synthesis of the first dinucleotide of RNA. The binding of the RNA polymerase to the DNA template is not affected. The example of sulfa drugs raises the important point that not all antibiotics are specific enough to be used in patients. Take the compound Actinomycin D, which targets DNA during RNA synthesis. It binds to DNA at guanine (G) and cytosine (C) basepairs and selectively inhibits RNA synthesis. Unfortunately, it is not selective for bacterial DNA and so is not used for treatment of bacterial infections. However, these compounds are very useful in the laboratory setting. Interference with Protein Synthesis Figure 3: Basic elements of protein translation. After DNA has been transcribed into messenger RNA (mRNA), this message is translated into protein (Figure 3). The process requires a ribosome, the mRNA, and a second type of RNA called transfer RNA (tRNA). It is the ribosome that is the cellular machine responsible for making the protein and it does this through two amino acid sites known as the “A” and “P” sites. For example, the first amino acid of the protein is carried to the “P” site by a tRNA that corresponds to a three-nucleotide sequence, known as a codon, within the mRNA sequence, while the codon sequence in the “A” site determines the identity of the next amino acid to be incorporated into the growing protein. As amino acids are brought to the ribosome by various tRNAs they are attached together and eventually an entire protein is created. There are antibiotics that inhibit the translational activity of the ribosome at various steps of protein synthesis1. For example, puromycin is a non-selective inhibitor of protein synthesis that is a mimic tRNA. It is incorporated into the ribosome at the “A” site and accepts the growing polypeptide chain by formation of a peptide bond, but it blocks the addition any more amino acids. Alternatively, streptomycin causes the incorporation of incorrect amino acids at the “A” site of the growing polypeptide, whereas, tetracyclines completely block protein translation by binding to a ribosomal subunit. The Evolution of Antibiotic Resistance There are resistance mechanisms for each of the antibiotics described above. Most often resistance results from either a change in a protein structure of the bacterium, an inactivation of the antibiotic drug, the prevention of antibiotic accumulation, or the block of its entry into a cell7. The increased development of “anti-antibiotic” strategies in bacterial cells is actually a result of the use of antibiotics. This is because the use of antibiotics creates a strong selective pressure that favors those bacteria that acquire such mechanisms of resistance. In a population of bacteria that are sensitive to an antibiotic, its use will prevent those bacteria from leaving descendant or daughter cells. However, in any population of bacteria there will be occasional random mutations in the protein sequence of the various enzymes within any particular cell. If one of these mutations gives rise to a protein that is impervious to the action of the antibiotic, that cell will survive and produce descendant cells that are also resistant to the activity of the antibiotic. In fact, the biology of bacteria provides ideal opportunities for these chance occurrences of resistance. Since, under ideal conditions an E. coli bacterium can divide every two hours, the chance it making a beneficial mistake is high enough for such resistances to occur and flourish due to the antibiotic selection. Random mutation is not the only way that a bacterial cell can acquire resistance to antibiotics. Bacteria can also take up foreign DNA from their environment and from other bacteria. Thus, if a bacterium of one species is resistant to an antibiotic it is possible that the DNA encoding the resistant protein may be transferred to bacteria of another (formerly sensitive) species. The acquisition of resistance in bacteria is a serious problem1,7. Some antibiotics are no longer useful for treating infections because bacterial resistance to the antibiotic has spread worldwide. Bacterial resistance to antibiotics can develop and spread very quickly, rendering an antibiotic ineffective within only a few years. The development of new antibiotics is both expensive and time-consuming and in some cases it appears that bacteria are developing resistance to antibiotics faster than scientists can develop them. It is hoped that our continued drive to understand how antibiotics work will keep these useful drugs available. Additional Reading 1. McDermott PF, et al Antimicrobials: Modes of Action and Mechanisms of Resistance. Int J Toxicol 22(2): Amyes S Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics. London/New York: Taylor & Francis. 262 p Chopra I, et al Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol 92 Suppl: 4S-15S Scott GM, Kyi MS Handbook of Essential Antibiotics. Amsterdam: Harwood Academic. 117p. References 1. Walsh C Antibiotics: Actions, Origins, Resistance. Washington, DC: ASM Press. 335p Birch B Alexander Fleming: The Bacteriologist who Discovered Penicillin, A Miracle Drug that has Saved Millions of Lives. Toronto: Irwin Pub. 64p A History of Antibiotics [videorecording]: A Presentation of Films for the Humanities & Sciences. Princeton, NJ: Films for the Humanities and Sciences, c videocassette (45 min.). 4. Beveridge TJ Use of the gram stain in microbiology. Biotech Histochem 76(3): Shockman GD, Barrett JF Structure, function, and assembly of cell walls of gram-positive bacteria. Annu Rev Microbiol 37: Ghuysen JM, et al Penicillin and beyond: evolution, protein fold, multimodular polypeptides, and multiprotein complexes. Microb Drug Resist 2(2): Walsh C Molecular mechanisms that confer antibacterial drug resistance. Nature 406(6797): Contact us: Related Articles Related Resources A related article reference A second reference a third reference A relevant teaching resource A second etc. Alexander Fleming, 1928, Penicilium Η.W. Florey και E.B. Chain 1941, Βιομ. παραγ. πενικιλλίνης Gerhard Domagk 1935, Prontosil 16
, Regional Research Laboratory in Peoria, Illinois became the first site for. commercial production of penicillin [5]. By mid-1944, when the Allies invaded. France, large supplies of the yellow liquid were available. When. treated with penicillin, 95% of the wounded lived. The Nobel Prize in Medicine. was awarded to Fleming, Florey, and Chain in Selman A. Waksman, a Russian immigrant to the United States, gave the. name ‘‘antibiotics’’ to chemicals (produced by soil-borne fungi and microorganisms) that destroy or slow the growth of other microbes. Waksman. spent his lifetime hunting for ‘‘antibiotic’’-producing micro-organisms and. in 1943 found a mold that was able to kill Tubercle bacilli. He called this. aminoglycoside streptomycin. On November 20, 1944, streptomycin was administered. to a young woman who had advanced pulmonary tuberculosis at. the Mayo Clinic in Rochester, Minnesota. Her life was saved by streptomycin. Dr. Waksman received the Nobel Prize in 1952 for its discovery. The rest is history: awakening to the possibilities and confronting. the challenges. Subsequently, more broad-spectrum penicillins and other aminoglycosides. were developed, followed by more antibiotic classes. More than antibiotics are now known. Approximately a thousand of these have been. carefully investigated, and about 100 are currently used to treat infections. Most are produced by actinomycetes and bacteria, many of which are. then chemically modified (semisynthetic). Others are completely synthetic. The b-lactams. Penicillin, the prototype b-lactam, is a 6-aminopenicillanic acid consisting. of a thiazolidine ring, an attached b-lactam ring, and a side chain [6]. Manipulations of the side chain have altered b-lactamase susceptibility, antibacterial spectrum, and pharmacokinetic properties. Other groups of. antibacterial agents that contain the b-lactam ring include cephalosporins, carbapenems, and monobactams. In actively dividing bacteria, the b-lactams. inhibit enzymes (transpeptidase, carboxypeptidase, and endopeptidase) located beneath the cell wall that are termed the ‘‘penicillin-binding. proteins.’’ This inhibition prevents the development of normal peptidoglycan. structure, because these enzymes are involved in creating the cross-linkage. between the peptide chains. Various bacteria differ in the permeability of. their cell walls to antibiotics and the type and concentration of penicillinbinding. proteins. Subsequent activation of the endogenous autolytic system. of bacteria by b-lactams initiates cell lysis and death [7]. Penicillins. The penicillin family of antibiotics remains an important part of today’s. antimicrobial armamentarium. Penicillins are bactericidal against most. ANTIBIOTICS: PAST, PRESENT, FUTURE It was in the mid-nineteenth century that Louis Pasteur observed that. some micro-organisms destroy othersdthe phenomenon that later came. to be known as antibiosis or ‘‘against life.’’ The search for antimicrobial. chemical agents revealed that antiseptics were too toxic for anything but surface. use on wounds. German bacteriologist Paul Ehrlich systematically. tested chemical agents, searching for the ‘‘magic bullet’’ that could be taken. internally, but he ended up with only a high-risk arsenic-based treatment for. syphilis. Alexander Fleming in London had been looking for antibacterial. agents in human secretions. The discovery of the antibacterial activity of. the enzyme lysozyme was made because of an accidental sneeze on a petri. dish [3]. Fleming observed that, when bacteria later formed colonies on. the plate, none developed in the spots occupied by mucus. Further tests. showed that lysozyme acted mostly against harmless organisms. In 1928, serendipity made another notable visit to Fleming’s laboratory at St. Mary’s. Hospital in London. He had left a culture plate of Staphylococci uncovered. in his laboratory while on a vacation. On his return, he noticed mold in the. petri dish along with a clear space between the Staphylococci and the bluegreen. spotted mold. It was the classic example of what Pasteur had referred. to as fortune’s accommodating a willing mind. Fleming identified the mold as Penicillium natatum, a culture filtrate of. which was able to kill bacteria. He named the agent in the filtrate penicillin. Because of a paucity of financial resources and Fleming’s modest ambitions. (according to some historians), it took another 12 years for penicillin to. emerge as the greatest medical advance of the twentieth (or any other) century. But the golden age of anti-infective medicine actually began in Gerhard Domagk, a German pharmacologist, discovered that a dye used. to tint cloth cured streptococcal infections in mice. His own dying daughter. survived a streptococcal infection after he injected her with the dye. Daniel. Bovert, a Swiss-born scientist, identified the active compound as sulfanilamide. Domagk was awarded the Nobel Prize in Medicine in Florey and Chain, working at Oxford University, were interested to note. that staphylococci, though resistant to sulfanamides and lysozyme, were apparently. sensitive to the penicillium mold [4]. World War II provided a crucial. spur to and much-needed resources for research on antimicrobial. agents. Staphylococcal infections and gas gangrene were killing more men. than the immediate organ damage caused by shell and bullet wounds. In. the spring of 1940, Florey and Chain were able to make a small amount. of yellowish-brown powder from Fleming’s mold. This first sample of ‘‘penicillin. powder’’ was a million times more potent than Fleming’s original filtrate. In 1941, the Fermentation Division of the newly created Northern Regional Research Laboratory in Peoria, Illinois became the first site for. Dr. Waksman received the Nobel Prize in 1952 for its discovery.Antibiotics are compounds that act to kill or inhibit the growth of bacteria1. The etymology of the term can be broken down into two roots: the prefix anti- meaning opposed to or preventing and biotic coming from the Greek word for life. In nature, various microbes and fungi secrete these compounds to gain an advantage in their microenvironment and it is from these very organisms that antibiotics are commonly use isolated1. The Discovery of Antibiotics. The stories of the discovery of antibiotics are dramatic and full of human interest, both on a global and personal scale. Two brief recounts are given below (please see reference 2 for an excellent overview of antibiotics). Alexander Fleming is popularly thought to have been the discoverer of penicillin. He is certainly the first researcher to have recognized its potential. In 1928 Fleming discovered that a blue mold (Penicillium) was able to lyse bacterial Staphylococci cells. Fleming determined that Penicillium produced some compound that caused the bacterial cells to lyse. He called this compound penicillin. And how he came to these conclusions is a now famous story of fortuitous chance3. Fleming had returned to the lab after a holiday to discover that some culture plates of Staphylococci had become contaminated. It was an accidental growth of Penicillium, but luckily one he did not throw away. His further observation that the contaminating mold was able to kill Staphylococci led to his being awarded the Nobel Prize for Medicine in Howard Florey and Ernest Chain were also honoured with the Nobel Prize for developing a way to produce large quantities of penicillin. Gerhard Domagk discovered the first sulfa drugs in The pharmaceutical company Bayer had hired him to work on the problem of infectious diseases caused by the bacterium Streptococcus pyogenes. In various trials designed to determine the effectiveness of various compounds for bacterial killing, Domagk discovered that a dye called prontosil rubrum prevented S. pyogenes infection in mice. In 1935 Domagk’s daughter was gravely ill due to S. pyogenes infection. The infection was advancing so aggressively that doctors were considering amputating her arm, but instead she was treated with prontosil rubrum (well before complete clinical trials of the drug had been completed). She made a full recovery. Domagk received the Nobel Prize in Mechanism of Action. Being such an important medical compound the mechanisms by which antibiotics kill bacteria have been under scrutiny for decades, with such studies being instrumental in the design of new and improved compounds. There are three general modes of antibiotic activity: (1) interference with the cell wall, (2) interference with nucleic acid synthesis, and (3) interference with protein synthesis. Figure 1. A basic schematic showing the arrangement of the cell wall in relation to the plasma membrane of a bacterial cell. The thickness and composition of the cell wall is different between the Gram-positive and Gram-negative cells. Interference with the Cell Wall. There is a multitude of ways to classify bacteria, but one of the more common methods is as either Gram-positive or Gram-negative cells4. These Gram designations are based on a differential staining assay, with the bacteria that stain dark being referred to as Gram-positive, while those that do not stain dark being referred to as Gram-negative. This difference in stain intensity and thus designation does have a physical basis, which is linked to the cell membrane. In every bacterial cell the plasma membrane encases the contents of the cell (referred to as the cytoplasm) and directly outside the plasma membrane is an additional exterior cell wall (See Figure 1). The plasma membrane of the cell is pressed tightly against the cell wall due to turgor pressure. It is the cell wall that determines the shape of the cells, the strength of which is provided by peptidoglycan5 (the major structural component of the cell wall). Gram-positive bacteria have thick cell walls composed primarily of this substance; where as Gram-negative bacteria have multi-layer cell walls that are thinner than those of the Gram-positive cells. Peptidoglycan is a lattice-like macromolecule composed of repeated sugar units that are cross-linked together, with the glycans of peptidoglycan forming polysaccharide strands that run parallel to each other (See Figure 2). These polysaccharide strands are composed of alternating units of two sugars: N-acetylmuramic acid (commonly referred to as NAM) and N-acetylglucosamine (commonly referred to as NAG). Additionally, peptidoglycan contains two peptides that cross-link these long polysaccharide strands and are composed of alanine, glutamic acid, lysine and alanine. Tetra-peptides linking the polysaccharide strands are themselves linked by a penta-peptide of five glycine residues that runs from the lysine residue of one tetrapeptide to the terminal alanine residue of another tetra-peptide. Figure 2 shows how these three different components (the polysaccharide strands, the tetra-peptide and the penta-peptide) come together to form peptidoglycan and give this macromoleclue its strength. Figure 2. A schematic of peptidoglycan’s structure. The NAM and NAG sugars are shown as green and blue spheres respectively. The tetrapeptides linked to NAM are cross-linked by a pentaglycine peptide, shown as red lines linking the D-glutamine (L) to the D-alanine (A).] Peptidoglycan is synthesized in three stages in three different parts of the cell. Firstly, the NAM sugar is linked to the alanine, glutamic acid, lysine and alanine precursor in the cytoplasm, forming the basic subunit of peptidoglycan. Secondly, the NAM/peptide is linked to the NAG sugar at the cell membrane. And lastly, now at the cell wall, the newly synthesized peptidoglycan subunit is transferred to the growing point of the cell wall’s peptidoglycan by a bond between the old peptidoglycan and the new NAM-NAG disaccharide. Here the lateral cross-linking by the pentaglycine peptide can occur. Antibiotics are active at every step of peptidoglycan synthesis. Among the most famous of the antibiotics to interfere with peptidoglycan synthesis are the b-lactams, of which penicillin is an example6. Penicillins resemble the terminal amino acids of the NAM+tetrapeptide precursor synthesized in the cytoplasm and actually bind the enzyme that catalyzes the pentaglycine cross-linking reaction. This binding prevents the cross-linking reaction from occurring and thus weakens the cell wall. Eventually, the turgor pressure of the cell causes the cell to lose its shape and eventually burst if the surrounding solution is hypotonic. Go to to see a short movie showing the activity of penicillin on Escherichia coli. On the flip side of this coin, bacteria can become resistant to penicillin by three strategies: the hydrolysis of penicillin, the acquisition of proteins with a reduced affinity for penicillin or the reduced uptake of penicillin. The hydrolysis of penicillin is catalyzed by enzymes called b-lactamases, which ring structure of penicillin and prevent it from mimicing the structure of the peptidoglycan precursor. Interference with Nucleic Acid Synthesis. Antibiotics are frequently active against nucleic acid synthesis within the bacterial cell1. This includes inhibition or interference of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis. The antibiotics that act in this manner are frequently analogs of essential metabolites of the cell, compounds that block DNA template activity during synthesis of new DNA, or compounds that block the transcription of DNA into RNA An example is the sulfonamides, commonly known as sulfa drugs, which are derivatives of dyes and resemble the compound folate. Folate is a coenzyme (a substance required for the proper functioning of enzymes) that is essential for cell growth and in bacterial cells is a precursor for the synthesis of amino acids and nucleic acids. Most bacteria synthesize folate from scratch, whereas mammalian cells cannot do this and must transport folate, which has been made by other sources, into their cells. This metabolic difference between bacterial cells and mammalian cells makes folate biosynthesis a convenient target for antibiotics, as this pathway is specific to bacterial cells. The sulfa drugs mimic one of the folate precursors, competing with the precursor for the enzyme involved in the next step of folate synthesis. If the normal folate precursor cannot bind the enzyme (because the sulfa drug is there), folate synthesis is blocked. Thus, the bacteria cannot synthesize the nucleic acids and some of the amino acids necessary for cell survival and perish. The method by which an antibiotic affects nucleic acid synthesis is actually quite diverse. For example, the coiling of the bacterial chromosome can be attacked. It is quite a long structure and its great size requires that this it is efficiently packed into the cell by supercoiling. The tightly coiled DNA must be uncoiled and relaxed in order for the DNA or RNA polymerases to gain access to the DNA template, a process accomplished by enzymes known as topoisomerases. All three types of enzymes, the RNA and DNA polymerases and topoisomerases, are antibiotic targets. Drugs called quinolones target a topoisomerase known as ‘DNA gyrase’, which normally uncoils DNA by cutting the two strands and then passing a section of the double helix through the gap. Quinolones bind to the cut strands of DNA, preventing the re-annealing (gluing back together) of the cut DNA strands with the parent strands. An alternative is the nitroimidazoles, drugs that cleave the double-stranded DNA template by producing radical ions. Cleavage of the DNA template interferes with both DNA replication and RNA synthesis. Classes of drugs known as rifamycins actually specifically inhibit RNA synthesis by binding to the RNA polymerase and preventing the synthesis of the first dinucleotide of RNA. The binding of the RNA polymerase to the DNA template is not affected. The example of sulfa drugs raises the important point that not all antibiotics are specific enough to be used in patients. Take the compound Actinomycin D, which targets DNA during RNA synthesis. It binds to DNA at guanine (G) and cytosine (C) basepairs and selectively inhibits RNA synthesis. Unfortunately, it is not selective for bacterial DNA and so is not used for treatment of bacterial infections. However, these compounds are very useful in the laboratory setting. Interference with Protein Synthesis. Figure 3: Basic elements of protein translation. After DNA has been transcribed into messenger RNA (mRNA), this message is translated into protein (Figure 3). The process requires a ribosome, the mRNA, and a second type of RNA called transfer RNA (tRNA). It is the ribosome that is the cellular machine responsible for making the protein and it does this through two amino acid sites known as the A and P sites. For example, the first amino acid of the protein is carried to the P site by a tRNA that corresponds to a three-nucleotide sequence, known as a codon, within the mRNA sequence, while the codon sequence in the A site determines the identity of the next amino acid to be incorporated into the growing protein. As amino acids are brought to the ribosome by various tRNAs they are attached together and eventually an entire protein is created. There are antibiotics that inhibit the translational activity of the ribosome at various steps of protein synthesis1. For example, puromycin is a non-selective inhibitor of protein synthesis that is a mimic tRNA. It is incorporated into the ribosome at the A site and accepts the growing polypeptide chain by formation of a peptide bond, but it blocks the addition any more amino acids. Alternatively, streptomycin causes the incorporation of incorrect amino acids at the A site of the growing polypeptide, whereas, tetracyclines completely block protein translation by binding to a ribosomal subunit. The Evolution of Antibiotic Resistance. There are resistance mechanisms for each of the antibiotics described above. Most often resistance results from either a change in a protein structure of the bacterium, an inactivation of the antibiotic drug, the prevention of antibiotic accumulation, or the block of its entry into a cell7. The increased development of anti-antibiotic strategies in bacterial cells is actually a result of the use of antibiotics. This is because the use of antibiotics creates a strong selective pressure that favors those bacteria that acquire such mechanisms of resistance. In a population of bacteria that are sensitive to an antibiotic, its use will prevent those bacteria from leaving descendant or daughter cells. However, in any population of bacteria there will be occasional random mutations in the protein sequence of the various enzymes within any particular cell. If one of these mutations gives rise to a protein that is impervious to the action of the antibiotic, that cell will survive and produce descendant cells that are also resistant to the activity of the antibiotic. In fact, the biology of bacteria provides ideal opportunities for these chance occurrences of resistance. Since, under ideal conditions an E. coli bacterium can divide every two hours, the chance it making a beneficial mistake is high enough for such resistances to occur and flourish due to the antibiotic selection. Random mutation is not the only way that a bacterial cell can acquire resistance to antibiotics. Bacteria can also take up foreign DNA from their environment and from other bacteria. Thus, if a bacterium of one species is resistant to an antibiotic it is possible that the DNA encoding the resistant protein may be transferred to bacteria of another (formerly sensitive) species. The acquisition of resistance in bacteria is a serious problem1,7. Some antibiotics are no longer useful for treating infections because bacterial resistance to the antibiotic has spread worldwide. Bacterial resistance to antibiotics can develop and spread very quickly, rendering an antibiotic ineffective within only a few years. The development of new antibiotics is both expensive and time-consuming and in some cases it appears that bacteria are developing resistance to antibiotics faster than scientists can develop them. It is hoped that our continued drive to understand how antibiotics work will keep these useful drugs available. Additional Reading. 1. McDermott PF, et al Antimicrobials: Modes of Action and Mechanisms of Resistance. Int J Toxicol 22(2): Amyes S Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics. London/New York: Taylor & Francis. 262 p. 3. Chopra I, et al Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol 92 Suppl: 4S-15S. 4. Scott GM, Kyi MS Handbook of Essential Antibiotics. Amsterdam: Harwood Academic. 117p. References. 1. Walsh C Antibiotics: Actions, Origins, Resistance. Washington, DC: ASM Press. 335p. 2. Birch B Alexander Fleming: The Bacteriologist who Discovered Penicillin, A Miracle Drug that has Saved Millions of Lives. Toronto: Irwin Pub. 64p. 3. A History of Antibiotics [videorecording]: A Presentation of Films for the Humanities & Sciences. Princeton, NJ: Films for the Humanities and Sciences, c videocassette (45 min.). 4. Beveridge TJ Use of the gram stain in microbiology. Biotech Histochem 76(3): Shockman GD, Barrett JF Structure, function, and assembly of cell walls of gram-positive bacteria. Annu Rev Microbiol 37: Ghuysen JM, et al Penicillin and beyond: evolution, protein fold, multimodular polypeptides, and multiprotein complexes. Microb Drug Resist 2(2): Walsh C Molecular mechanisms that confer antibacterial drug resistance. Nature 406(6797): Contact us: Related Articles Related Resources. A related article reference A second reference a third reference A relevant teaching resource A second etc. Alexander Fleming, 1928, Penicilium. Η.W. Florey και E.B. Chain. 1941, Βιομ. παραγ. πενικιλλίνης. Gerhard Domagk. 1935, Prontosil. 16.")
17
17

19
Η ΑΝΑΚΑΛΥΨΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ
Selman Abraham Waksman (22 July 1888 -16 August 1973) Ανακάλυψε τη Στρεπτομυκίνη και άλλα αντιβιοτικά Nobel Prize,1952 It was in the mid-nineteenth century that Louis Pasteur observed that some micro-organisms destroy othersdthe phenomenon that later came to be known as antibiosis or ‘‘against life.’’ The search for antimicrobial chemical agents revealed that antiseptics were too toxic for anything but surface use on wounds. German bacteriologist Paul Ehrlich systematically tested chemical agents, searching for the ‘‘magic bullet’’ that could be taken internally, but he ended up with only a high-risk arsenic-based treatment for syphilis. Alexander Fleming in London had been looking for antibacterial agents in human secretions. The discovery of the antibacterial activity of the enzyme lysozyme was made because of an accidental sneeze on a petri dish [3]. Fleming observed that, when bacteria later formed colonies on the plate, none developed in the spots occupied by mucus. Further tests showed that lysozyme acted mostly against harmless organisms. In 1928, serendipity made another notable visit to Fleming’s laboratory at St. Mary’s Hospital in London. He had left a culture plate of Staphylococci uncovered in his laboratory while on a vacation. On his return, he noticed mold in the petri dish along with a clear space between the Staphylococci and the bluegreen spotted mold. It was the classic example of what Pasteur had referred to as fortune’s accommodating a willing mind. Fleming identified the mold as Penicillium natatum, a culture filtrate of which was able to kill bacteria. He named the agent in the filtrate penicillin. Because of a paucity of financial resources and Fleming’s modest ambitions (according to some historians), it took another 12 years for penicillin to emerge as the greatest medical advance of the twentieth (or any other) century. But the golden age of anti-infective medicine actually began in 1934. Gerhard Domagk, a German pharmacologist, discovered that a dye used to tint cloth cured streptococcal infections in mice. His own dying daughter survived a streptococcal infection after he injected her with the dye. Daniel Bovert, a Swiss-born scientist, identified the active compound as sulfanilamide. Domagk was awarded the Nobel Prize in Medicine in 1939. Florey and Chain, working at Oxford University, were interested to note that staphylococci, though resistant to sulfanamides and lysozyme, were apparently sensitive to the penicillium mold [4]. World War II provided a crucial spur to and much-needed resources for research on antimicrobial agents. Staphylococcal infections and gas gangrene were killing more men than the immediate organ damage caused by shell and bullet wounds. In the spring of 1940, Florey and Chain were able to make a small amount of yellowish-brown powder from Fleming’s mold. This first sample of ‘‘penicillin powder’’ was a million times more potent than Fleming’s original filtrate. In 1941, the Fermentation Division of the newly created Northern Regional Research Laboratory in Peoria, Illinois became the first site for commercial production of penicillin [5]. By mid-1944, when the Allies invaded France, large supplies of the yellow liquid were available. When treated with penicillin, 95% of the wounded lived. The Nobel Prize in Medicine was awarded to Fleming, Florey, and Chain in 1945. Selman A. Waksman, a Russian immigrant to the United States, gave the name ‘‘antibiotics’’ to chemicals (produced by soil-borne fungi and microorganisms) that destroy or slow the growth of other microbes. Waksman spent his lifetime hunting for ‘‘antibiotic’’-producing micro-organisms and in 1943 found a mold that was able to kill Tubercle bacilli. He called this aminoglycoside streptomycin. On November 20, 1944, streptomycin was administered to a young woman who had advanced pulmonary tuberculosis at the Mayo Clinic in Rochester, Minnesota. Her life was saved by streptomycin. Dr. Waksman received the Nobel Prize in 1952 for its discovery. Antibiotics are compounds that act to kill or inhibit the growth of bacteria1. The etymology of the term can be broken down into two roots: the prefix “anti-” meaning “opposed to” or “preventing” and “biotic” coming from the Greek word for life. In nature, various microbes and fungi secrete these compounds to gain an advantage in their microenvironment and it is from these very organisms that antibiotics are commonly use isolated1. The Discovery of Antibiotics The stories of the discovery of antibiotics are dramatic and full of human interest, both on a global and personal scale. Two brief recounts are given below (please see reference 2 for an excellent overview of antibiotics). Alexander Fleming is popularly thought to have been the discoverer of penicillin. He is certainly the first researcher to have recognized its potential. In 1928 Fleming discovered that a blue mold (Penicillium) was able to lyse bacterial Staphylococci cells. Fleming determined that Penicillium produced some compound that caused the bacterial cells to lyse. He called this compound penicillin. And how he came to these conclusions is a now famous story of fortuitous chance3. Fleming had returned to the lab after a holiday to discover that some culture plates of Staphylococci had become contaminated. It was an accidental growth of Penicillium, but luckily one he did not throw away. His further observation that the contaminating mold was able to kill Staphylococci led to his being awarded the Nobel Prize for Medicine in Howard Florey and Ernest Chain were also honoured with the Nobel Prize for developing a way to produce large quantities of penicillin. Gerhard Domagk discovered the first sulfa drugs in The pharmaceutical company Bayer had hired him to work on the problem of infectious diseases caused by the bacterium Streptococcus pyogenes. In various trials designed to determine the effectiveness of various compounds for bacterial killing, Domagk discovered that a dye called prontosil rubrum prevented S. pyogenes infection in mice. In 1935 Domagk’s daughter was gravely ill due to S. pyogenes infection. The infection was advancing so aggressively that doctors were considering amputating her arm, but instead she was treated with prontosil rubrum (well before complete clinical trials of the drug had been completed). She made a full recovery. Domagk received the Nobel Prize in Mechanism of Action Being such an important medical compound the mechanisms by which antibiotics kill bacteria have been under scrutiny for decades, with such studies being instrumental in the design of new and improved compounds. There are three general modes of antibiotic activity: (1) interference with the cell wall, (2) interference with nucleic acid synthesis, and (3) interference with protein synthesis Figure 1. A basic schematic showing the arrangement of the cell wall in relation to the plasma membrane of a bacterial cell. The thickness and composition of the cell wall is different between the Gram-positive and Gram-negative cells. Interference with the Cell Wall There is a multitude of ways to classify bacteria, but one of the more common methods is as either Gram-positive or Gram-negative cells4. These Gram designations are based on a differential staining assay, with the bacteria that stain dark being referred to as Gram-positive, while those that do not stain dark being referred to as Gram-negative. This difference in stain intensity and thus designation does have a physical basis, which is linked to the cell membrane. In every bacterial cell the plasma membrane encases the contents of the cell (referred to as the cytoplasm) and directly outside the plasma membrane is an additional exterior cell wall (See Figure 1). The plasma membrane of the cell is pressed tightly against the cell wall due to turgor pressure. It is the cell wall that determines the shape of the cells, the strength of which is provided by peptidoglycan5 (the major structural component of the cell wall). Gram-positive bacteria have thick cell walls composed primarily of this substance; where as Gram-negative bacteria have multi-layer cell walls that are thinner than those of the Gram-positive cells. Peptidoglycan is a lattice-like macromolecule composed of repeated sugar units that are cross-linked together, with the “glycans” of peptidoglycan forming polysaccharide strands that run parallel to each other (See Figure 2). These polysaccharide strands are composed of alternating units of two sugars: N-acetylmuramic acid (commonly referred to as NAM) and N-acetylglucosamine (commonly referred to as NAG). Additionally, peptidoglycan contains two peptides that cross-link these long polysaccharide strands and are composed of alanine, glutamic acid, lysine and alanine. Tetra-peptides linking the polysaccharide strands are themselves linked by a penta-peptide of five glycine residues that runs from the lysine residue of one tetrapeptide to the terminal alanine residue of another tetra-peptide. Figure 2 shows how these three different components (the polysaccharide strands, the tetra-peptide and the penta-peptide) come together to form peptidoglycan and give this macromoleclue its strength Figure 2. A schematic of peptidoglycan’s structure. The NAM and NAG sugars are shown as green and blue spheres respectively. The tetrapeptides linked to NAM are cross-linked by a pentaglycine peptide, shown as red lines linking the D-glutamine (L) to the D-alanine (A).] Peptidoglycan is synthesized in three stages in three different parts of the cell. Firstly, the NAM sugar is linked to the alanine, glutamic acid, lysine and alanine precursor in the cytoplasm, forming the basic subunit of peptidoglycan. Secondly, the NAM/peptide is linked to the NAG sugar at the cell membrane. And lastly, now at the cell wall, the newly synthesized peptidoglycan subunit is transferred to the growing point of the cell wall’s peptidoglycan by a bond between the old peptidoglycan and the new NAM-NAG disaccharide. Here the lateral cross-linking by the pentaglycine peptide can occur. Antibiotics are active at every step of peptidoglycan synthesis. Among the most famous of the antibiotics to interfere with peptidoglycan synthesis are the b-lactams, of which penicillin is an example6. Penicillins resemble the terminal amino acids of the NAM+tetrapeptide precursor synthesized in the cytoplasm and actually bind the enzyme that catalyzes the pentaglycine cross-linking reaction. This binding prevents the cross-linking reaction from occurring and thus weakens the cell wall. Eventually, the turgor pressure of the cell causes the cell to lose its shape and eventually burst if the surrounding solution is hypotonic. Go to to see a short movie showing the activity of penicillin on Escherichia coli. On the flip side of this coin, bacteria can become resistant to penicillin by three strategies: the hydrolysis of penicillin, the acquisition of proteins with a reduced affinity for penicillin or the reduced uptake of penicillin. The hydrolysis of penicillin is catalyzed by enzymes called b-lactamases, which ring structure of penicillin and prevent it from mimicing the structure of the peptidoglycan precursor. Interference with Nucleic Acid Synthesis Antibiotics are frequently active against nucleic acid synthesis within the bacterial cell1. This includes inhibition or interference of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis. The antibiotics that act in this manner are frequently analogs of essential metabolites of the cell, compounds that block DNA template activity during synthesis of new DNA, or compounds that block the transcription of DNA into RNA An example is the sulfonamides, commonly known as sulfa drugs, which are derivatives of dyes and resemble the compound folate. Folate is a coenzyme (a substance required for the proper functioning of enzymes) that is essential for cell growth and in bacterial cells is a precursor for the synthesis of amino acids and nucleic acids. Most bacteria synthesize folate from scratch, whereas mammalian cells cannot do this and must transport folate, which has been made by other sources, into their cells. This metabolic difference between bacterial cells and mammalian cells makes folate biosynthesis a convenient target for antibiotics, as this pathway is specific to bacterial cells. The sulfa drugs mimic one of the folate precursors, competing with the precursor for the enzyme involved in the next step of folate synthesis. If the normal folate precursor cannot bind the enzyme (because the sulfa drug is there), folate synthesis is blocked. Thus, the bacteria cannot synthesize the nucleic acids and some of the amino acids necessary for cell survival and perish. The method by which an antibiotic affects nucleic acid synthesis is actually quite diverse. For example, the coiling of the bacterial chromosome can be attacked. It is quite a long structure and its great size requires that this it is efficiently packed into the cell by supercoiling. The tightly coiled DNA must be uncoiled and relaxed in order for the DNA or RNA polymerases to gain access to the DNA template, a process accomplished by enzymes known as topoisomerases. All three types of enzymes, the RNA and DNA polymerases and topoisomerases, are antibiotic targets. Drugs called quinolones target a topoisomerase known as ‘DNA gyrase’, which normally uncoils DNA by cutting the two strands and then passing a section of the double helix through the gap. Quinolones bind to the cut strands of DNA, preventing the re-annealing (gluing back together) of the cut DNA strands with the parent strands. An alternative is the nitroimidazoles, drugs that cleave the double-stranded DNA template by producing radical ions. Cleavage of the DNA template interferes with both DNA replication and RNA synthesis. Classes of drugs known as rifamycins actually specifically inhibit RNA synthesis by binding to the RNA polymerase and preventing the synthesis of the first dinucleotide of RNA. The binding of the RNA polymerase to the DNA template is not affected. The example of sulfa drugs raises the important point that not all antibiotics are specific enough to be used in patients. Take the compound Actinomycin D, which targets DNA during RNA synthesis. It binds to DNA at guanine (G) and cytosine (C) basepairs and selectively inhibits RNA synthesis. Unfortunately, it is not selective for bacterial DNA and so is not used for treatment of bacterial infections. However, these compounds are very useful in the laboratory setting. Interference with Protein Synthesis Figure 3: Basic elements of protein translation. After DNA has been transcribed into messenger RNA (mRNA), this message is translated into protein (Figure 3). The process requires a ribosome, the mRNA, and a second type of RNA called transfer RNA (tRNA). It is the ribosome that is the cellular machine responsible for making the protein and it does this through two amino acid sites known as the “A” and “P” sites. For example, the first amino acid of the protein is carried to the “P” site by a tRNA that corresponds to a three-nucleotide sequence, known as a codon, within the mRNA sequence, while the codon sequence in the “A” site determines the identity of the next amino acid to be incorporated into the growing protein. As amino acids are brought to the ribosome by various tRNAs they are attached together and eventually an entire protein is created. There are antibiotics that inhibit the translational activity of the ribosome at various steps of protein synthesis1. For example, puromycin is a non-selective inhibitor of protein synthesis that is a mimic tRNA. It is incorporated into the ribosome at the “A” site and accepts the growing polypeptide chain by formation of a peptide bond, but it blocks the addition any more amino acids. Alternatively, streptomycin causes the incorporation of incorrect amino acids at the “A” site of the growing polypeptide, whereas, tetracyclines completely block protein translation by binding to a ribosomal subunit. The Evolution of Antibiotic Resistance There are resistance mechanisms for each of the antibiotics described above. Most often resistance results from either a change in a protein structure of the bacterium, an inactivation of the antibiotic drug, the prevention of antibiotic accumulation, or the block of its entry into a cell7. The increased development of “anti-antibiotic” strategies in bacterial cells is actually a result of the use of antibiotics. This is because the use of antibiotics creates a strong selective pressure that favors those bacteria that acquire such mechanisms of resistance. In a population of bacteria that are sensitive to an antibiotic, its use will prevent those bacteria from leaving descendant or daughter cells. However, in any population of bacteria there will be occasional random mutations in the protein sequence of the various enzymes within any particular cell. If one of these mutations gives rise to a protein that is impervious to the action of the antibiotic, that cell will survive and produce descendant cells that are also resistant to the activity of the antibiotic. In fact, the biology of bacteria provides ideal opportunities for these chance occurrences of resistance. Since, under ideal conditions an E. coli bacterium can divide every two hours, the chance it making a beneficial mistake is high enough for such resistances to occur and flourish due to the antibiotic selection. Random mutation is not the only way that a bacterial cell can acquire resistance to antibiotics. Bacteria can also take up foreign DNA from their environment and from other bacteria. Thus, if a bacterium of one species is resistant to an antibiotic it is possible that the DNA encoding the resistant protein may be transferred to bacteria of another (formerly sensitive) species. The acquisition of resistance in bacteria is a serious problem1,7. Some antibiotics are no longer useful for treating infections because bacterial resistance to the antibiotic has spread worldwide. Bacterial resistance to antibiotics can develop and spread very quickly, rendering an antibiotic ineffective within only a few years. The development of new antibiotics is both expensive and time-consuming and in some cases it appears that bacteria are developing resistance to antibiotics faster than scientists can develop them. It is hoped that our continued drive to understand how antibiotics work will keep these useful drugs available. Additional Reading 1. McDermott PF, et al Antimicrobials: Modes of Action and Mechanisms of Resistance. Int J Toxicol 22(2): Amyes S Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics. London/New York: Taylor & Francis. 262 p Chopra I, et al Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol 92 Suppl: 4S-15S Scott GM, Kyi MS Handbook of Essential Antibiotics. Amsterdam: Harwood Academic. 117p. References 1. Walsh C Antibiotics: Actions, Origins, Resistance. Washington, DC: ASM Press. 335p Birch B Alexander Fleming: The Bacteriologist who Discovered Penicillin, A Miracle Drug that has Saved Millions of Lives. Toronto: Irwin Pub. 64p A History of Antibiotics [videorecording]: A Presentation of Films for the Humanities & Sciences. Princeton, NJ: Films for the Humanities and Sciences, c videocassette (45 min.). 4. Beveridge TJ Use of the gram stain in microbiology. Biotech Histochem 76(3): Shockman GD, Barrett JF Structure, function, and assembly of cell walls of gram-positive bacteria. Annu Rev Microbiol 37: Ghuysen JM, et al Penicillin and beyond: evolution, protein fold, multimodular polypeptides, and multiprotein complexes. Microb Drug Resist 2(2): Walsh C Molecular mechanisms that confer antibacterial drug resistance. Nature 406(6797): Contact us: Related Articles Related Resources A related article reference A second reference a third reference A relevant teaching resource A second etc. 19
 Ανακάλυψε τη Στρεπτομυκίνη και άλλα αντιβιοτικά Nobel Prize,1952. It was in the mid-nineteenth century that Louis Pasteur observed that. some micro-organisms destroy othersdthe phenomenon that later came. to be known as antibiosis or ‘‘against life.’’ The search for antimicrobial. chemical agents revealed that antiseptics were too toxic for anything but surface. use on wounds. German bacteriologist Paul Ehrlich systematically. tested chemical agents, searching for the ‘‘magic bullet’’ that could be taken. internally, but he ended up with only a high-risk arsenic-based treatment for. syphilis. Alexander Fleming in London had been looking for antibacterial. agents in human secretions. The discovery of the antibacterial activity of. the enzyme lysozyme was made because of an accidental sneeze on a petri. dish [3]. Fleming observed that, when bacteria later formed colonies on. the plate, none developed in the spots occupied by mucus. Further tests. showed that lysozyme acted mostly against harmless organisms. In 1928, serendipity made another notable visit to Fleming’s laboratory at St. Mary’s. Hospital in London. He had left a culture plate of Staphylococci uncovered. in his laboratory while on a vacation. On his return, he noticed mold in the. petri dish along with a clear space between the Staphylococci and the bluegreen. spotted mold. It was the classic example of what Pasteur had referred. to as fortune’s accommodating a willing mind. Fleming identified the mold as Penicillium natatum, a culture filtrate of. which was able to kill bacteria. He named the agent in the filtrate penicillin. Because of a paucity of financial resources and Fleming’s modest ambitions. (according to some historians), it took another 12 years for penicillin to. emerge as the greatest medical advance of the twentieth (or any other) century. But the golden age of anti-infective medicine actually began in Gerhard Domagk, a German pharmacologist, discovered that a dye used. to tint cloth cured streptococcal infections in mice. His own dying daughter. survived a streptococcal infection after he injected her with the dye. Daniel. Bovert, a Swiss-born scientist, identified the active compound as sulfanilamide. Domagk was awarded the Nobel Prize in Medicine in Florey and Chain, working at Oxford University, were interested to note. that staphylococci, though resistant to sulfanamides and lysozyme, were apparently. sensitive to the penicillium mold [4]. World War II provided a crucial. spur to and much-needed resources for research on antimicrobial. agents. Staphylococcal infections and gas gangrene were killing more men. than the immediate organ damage caused by shell and bullet wounds. In. the spring of 1940, Florey and Chain were able to make a small amount. of yellowish-brown powder from Fleming’s mold. This first sample of ‘‘penicillin. powder’’ was a million times more potent than Fleming’s original filtrate. In 1941, the Fermentation Division of the newly created Northern Regional Research Laboratory in Peoria, Illinois became the first site for. commercial production of penicillin [5]. By mid-1944, when the Allies invaded. France, large supplies of the yellow liquid were available. When. treated with penicillin, 95% of the wounded lived. The Nobel Prize in Medicine. was awarded to Fleming, Florey, and Chain in Selman A. Waksman, a Russian immigrant to the United States, gave the. name ‘‘antibiotics’’ to chemicals (produced by soil-borne fungi and microorganisms) that destroy or slow the growth of other microbes. Waksman. spent his lifetime hunting for ‘‘antibiotic’’-producing micro-organisms and. in 1943 found a mold that was able to kill Tubercle bacilli. He called this. aminoglycoside streptomycin. On November 20, 1944, streptomycin was administered. to a young woman who had advanced pulmonary tuberculosis at. the Mayo Clinic in Rochester, Minnesota. Her life was saved by streptomycin. Dr. Waksman received the Nobel Prize in 1952 for its discovery. Antibiotics are compounds that act to kill or inhibit the growth of bacteria1. The etymology of the term can be broken down into two roots: the prefix anti- meaning opposed to or preventing and biotic coming from the Greek word for life. In nature, various microbes and fungi secrete these compounds to gain an advantage in their microenvironment and it is from these very organisms that antibiotics are commonly use isolated1. The Discovery of Antibiotics. The stories of the discovery of antibiotics are dramatic and full of human interest, both on a global and personal scale. Two brief recounts are given below (please see reference 2 for an excellent overview of antibiotics). Alexander Fleming is popularly thought to have been the discoverer of penicillin. He is certainly the first researcher to have recognized its potential. In 1928 Fleming discovered that a blue mold (Penicillium) was able to lyse bacterial Staphylococci cells. Fleming determined that Penicillium produced some compound that caused the bacterial cells to lyse. He called this compound penicillin. And how he came to these conclusions is a now famous story of fortuitous chance3. Fleming had returned to the lab after a holiday to discover that some culture plates of Staphylococci had become contaminated. It was an accidental growth of Penicillium, but luckily one he did not throw away. His further observation that the contaminating mold was able to kill Staphylococci led to his being awarded the Nobel Prize for Medicine in Howard Florey and Ernest Chain were also honoured with the Nobel Prize for developing a way to produce large quantities of penicillin. Gerhard Domagk discovered the first sulfa drugs in The pharmaceutical company Bayer had hired him to work on the problem of infectious diseases caused by the bacterium Streptococcus pyogenes. In various trials designed to determine the effectiveness of various compounds for bacterial killing, Domagk discovered that a dye called prontosil rubrum prevented S. pyogenes infection in mice. In 1935 Domagk’s daughter was gravely ill due to S. pyogenes infection. The infection was advancing so aggressively that doctors were considering amputating her arm, but instead she was treated with prontosil rubrum (well before complete clinical trials of the drug had been completed). She made a full recovery. Domagk received the Nobel Prize in Mechanism of Action. Being such an important medical compound the mechanisms by which antibiotics kill bacteria have been under scrutiny for decades, with such studies being instrumental in the design of new and improved compounds. There are three general modes of antibiotic activity: (1) interference with the cell wall, (2) interference with nucleic acid synthesis, and (3) interference with protein synthesis. Figure 1. A basic schematic showing the arrangement of the cell wall in relation to the plasma membrane of a bacterial cell. The thickness and composition of the cell wall is different between the Gram-positive and Gram-negative cells. Interference with the Cell Wall. There is a multitude of ways to classify bacteria, but one of the more common methods is as either Gram-positive or Gram-negative cells4. These Gram designations are based on a differential staining assay, with the bacteria that stain dark being referred to as Gram-positive, while those that do not stain dark being referred to as Gram-negative. This difference in stain intensity and thus designation does have a physical basis, which is linked to the cell membrane. In every bacterial cell the plasma membrane encases the contents of the cell (referred to as the cytoplasm) and directly outside the plasma membrane is an additional exterior cell wall (See Figure 1). The plasma membrane of the cell is pressed tightly against the cell wall due to turgor pressure. It is the cell wall that determines the shape of the cells, the strength of which is provided by peptidoglycan5 (the major structural component of the cell wall). Gram-positive bacteria have thick cell walls composed primarily of this substance; where as Gram-negative bacteria have multi-layer cell walls that are thinner than those of the Gram-positive cells. Peptidoglycan is a lattice-like macromolecule composed of repeated sugar units that are cross-linked together, with the glycans of peptidoglycan forming polysaccharide strands that run parallel to each other (See Figure 2). These polysaccharide strands are composed of alternating units of two sugars: N-acetylmuramic acid (commonly referred to as NAM) and N-acetylglucosamine (commonly referred to as NAG). Additionally, peptidoglycan contains two peptides that cross-link these long polysaccharide strands and are composed of alanine, glutamic acid, lysine and alanine. Tetra-peptides linking the polysaccharide strands are themselves linked by a penta-peptide of five glycine residues that runs from the lysine residue of one tetrapeptide to the terminal alanine residue of another tetra-peptide. Figure 2 shows how these three different components (the polysaccharide strands, the tetra-peptide and the penta-peptide) come together to form peptidoglycan and give this macromoleclue its strength. Figure 2. A schematic of peptidoglycan’s structure. The NAM and NAG sugars are shown as green and blue spheres respectively. The tetrapeptides linked to NAM are cross-linked by a pentaglycine peptide, shown as red lines linking the D-glutamine (L) to the D-alanine (A).] Peptidoglycan is synthesized in three stages in three different parts of the cell. Firstly, the NAM sugar is linked to the alanine, glutamic acid, lysine and alanine precursor in the cytoplasm, forming the basic subunit of peptidoglycan. Secondly, the NAM/peptide is linked to the NAG sugar at the cell membrane. And lastly, now at the cell wall, the newly synthesized peptidoglycan subunit is transferred to the growing point of the cell wall’s peptidoglycan by a bond between the old peptidoglycan and the new NAM-NAG disaccharide. Here the lateral cross-linking by the pentaglycine peptide can occur. Antibiotics are active at every step of peptidoglycan synthesis. Among the most famous of the antibiotics to interfere with peptidoglycan synthesis are the b-lactams, of which penicillin is an example6. Penicillins resemble the terminal amino acids of the NAM+tetrapeptide precursor synthesized in the cytoplasm and actually bind the enzyme that catalyzes the pentaglycine cross-linking reaction. This binding prevents the cross-linking reaction from occurring and thus weakens the cell wall. Eventually, the turgor pressure of the cell causes the cell to lose its shape and eventually burst if the surrounding solution is hypotonic. Go to to see a short movie showing the activity of penicillin on Escherichia coli. On the flip side of this coin, bacteria can become resistant to penicillin by three strategies: the hydrolysis of penicillin, the acquisition of proteins with a reduced affinity for penicillin or the reduced uptake of penicillin. The hydrolysis of penicillin is catalyzed by enzymes called b-lactamases, which ring structure of penicillin and prevent it from mimicing the structure of the peptidoglycan precursor. Interference with Nucleic Acid Synthesis. Antibiotics are frequently active against nucleic acid synthesis within the bacterial cell1. This includes inhibition or interference of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis. The antibiotics that act in this manner are frequently analogs of essential metabolites of the cell, compounds that block DNA template activity during synthesis of new DNA, or compounds that block the transcription of DNA into RNA An example is the sulfonamides, commonly known as sulfa drugs, which are derivatives of dyes and resemble the compound folate. Folate is a coenzyme (a substance required for the proper functioning of enzymes) that is essential for cell growth and in bacterial cells is a precursor for the synthesis of amino acids and nucleic acids. Most bacteria synthesize folate from scratch, whereas mammalian cells cannot do this and must transport folate, which has been made by other sources, into their cells. This metabolic difference between bacterial cells and mammalian cells makes folate biosynthesis a convenient target for antibiotics, as this pathway is specific to bacterial cells. The sulfa drugs mimic one of the folate precursors, competing with the precursor for the enzyme involved in the next step of folate synthesis. If the normal folate precursor cannot bind the enzyme (because the sulfa drug is there), folate synthesis is blocked. Thus, the bacteria cannot synthesize the nucleic acids and some of the amino acids necessary for cell survival and perish. The method by which an antibiotic affects nucleic acid synthesis is actually quite diverse. For example, the coiling of the bacterial chromosome can be attacked. It is quite a long structure and its great size requires that this it is efficiently packed into the cell by supercoiling. The tightly coiled DNA must be uncoiled and relaxed in order for the DNA or RNA polymerases to gain access to the DNA template, a process accomplished by enzymes known as topoisomerases. All three types of enzymes, the RNA and DNA polymerases and topoisomerases, are antibiotic targets. Drugs called quinolones target a topoisomerase known as ‘DNA gyrase’, which normally uncoils DNA by cutting the two strands and then passing a section of the double helix through the gap. Quinolones bind to the cut strands of DNA, preventing the re-annealing (gluing back together) of the cut DNA strands with the parent strands. An alternative is the nitroimidazoles, drugs that cleave the double-stranded DNA template by producing radical ions. Cleavage of the DNA template interferes with both DNA replication and RNA synthesis. Classes of drugs known as rifamycins actually specifically inhibit RNA synthesis by binding to the RNA polymerase and preventing the synthesis of the first dinucleotide of RNA. The binding of the RNA polymerase to the DNA template is not affected. The example of sulfa drugs raises the important point that not all antibiotics are specific enough to be used in patients. Take the compound Actinomycin D, which targets DNA during RNA synthesis. It binds to DNA at guanine (G) and cytosine (C) basepairs and selectively inhibits RNA synthesis. Unfortunately, it is not selective for bacterial DNA and so is not used for treatment of bacterial infections. However, these compounds are very useful in the laboratory setting. Interference with Protein Synthesis. Figure 3: Basic elements of protein translation. After DNA has been transcribed into messenger RNA (mRNA), this message is translated into protein (Figure 3). The process requires a ribosome, the mRNA, and a second type of RNA called transfer RNA (tRNA). It is the ribosome that is the cellular machine responsible for making the protein and it does this through two amino acid sites known as the A and P sites. For example, the first amino acid of the protein is carried to the P site by a tRNA that corresponds to a three-nucleotide sequence, known as a codon, within the mRNA sequence, while the codon sequence in the A site determines the identity of the next amino acid to be incorporated into the growing protein. As amino acids are brought to the ribosome by various tRNAs they are attached together and eventually an entire protein is created. There are antibiotics that inhibit the translational activity of the ribosome at various steps of protein synthesis1. For example, puromycin is a non-selective inhibitor of protein synthesis that is a mimic tRNA. It is incorporated into the ribosome at the A site and accepts the growing polypeptide chain by formation of a peptide bond, but it blocks the addition any more amino acids. Alternatively, streptomycin causes the incorporation of incorrect amino acids at the A site of the growing polypeptide, whereas, tetracyclines completely block protein translation by binding to a ribosomal subunit. The Evolution of Antibiotic Resistance. There are resistance mechanisms for each of the antibiotics described above. Most often resistance results from either a change in a protein structure of the bacterium, an inactivation of the antibiotic drug, the prevention of antibiotic accumulation, or the block of its entry into a cell7. The increased development of anti-antibiotic strategies in bacterial cells is actually a result of the use of antibiotics. This is because the use of antibiotics creates a strong selective pressure that favors those bacteria that acquire such mechanisms of resistance. In a population of bacteria that are sensitive to an antibiotic, its use will prevent those bacteria from leaving descendant or daughter cells. However, in any population of bacteria there will be occasional random mutations in the protein sequence of the various enzymes within any particular cell. If one of these mutations gives rise to a protein that is impervious to the action of the antibiotic, that cell will survive and produce descendant cells that are also resistant to the activity of the antibiotic. In fact, the biology of bacteria provides ideal opportunities for these chance occurrences of resistance. Since, under ideal conditions an E. coli bacterium can divide every two hours, the chance it making a beneficial mistake is high enough for such resistances to occur and flourish due to the antibiotic selection. Random mutation is not the only way that a bacterial cell can acquire resistance to antibiotics. Bacteria can also take up foreign DNA from their environment and from other bacteria. Thus, if a bacterium of one species is resistant to an antibiotic it is possible that the DNA encoding the resistant protein may be transferred to bacteria of another (formerly sensitive) species. The acquisition of resistance in bacteria is a serious problem1,7. Some antibiotics are no longer useful for treating infections because bacterial resistance to the antibiotic has spread worldwide. Bacterial resistance to antibiotics can develop and spread very quickly, rendering an antibiotic ineffective within only a few years. The development of new antibiotics is both expensive and time-consuming and in some cases it appears that bacteria are developing resistance to antibiotics faster than scientists can develop them. It is hoped that our continued drive to understand how antibiotics work will keep these useful drugs available. Additional Reading. 1. McDermott PF, et al Antimicrobials: Modes of Action and Mechanisms of Resistance. Int J Toxicol 22(2): Amyes S Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics. London/New York: Taylor & Francis. 262 p. 3. Chopra I, et al Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol 92 Suppl: 4S-15S. 4. Scott GM, Kyi MS Handbook of Essential Antibiotics. Amsterdam: Harwood Academic. 117p. References. 1. Walsh C Antibiotics: Actions, Origins, Resistance. Washington, DC: ASM Press. 335p. 2. Birch B Alexander Fleming: The Bacteriologist who Discovered Penicillin, A Miracle Drug that has Saved Millions of Lives. Toronto: Irwin Pub. 64p. 3. A History of Antibiotics [videorecording]: A Presentation of Films for the Humanities & Sciences. Princeton, NJ: Films for the Humanities and Sciences, c videocassette (45 min.). 4. Beveridge TJ Use of the gram stain in microbiology. Biotech Histochem 76(3): Shockman GD, Barrett JF Structure, function, and assembly of cell walls of gram-positive bacteria. Annu Rev Microbiol 37: Ghuysen JM, et al Penicillin and beyond: evolution, protein fold, multimodular polypeptides, and multiprotein complexes. Microb Drug Resist 2(2): Walsh C Molecular mechanisms that confer antibacterial drug resistance. Nature 406(6797): Contact us: Related Articles Related Resources. A related article reference A second reference a third reference A relevant teaching resource A second etc. 19.")
20
20 According to IOM and FDA, only two new classes of
antibiotics have been developed in the past 30 years, and resistance to one class emerged even before FDA approved the drug. (See Table 2.) Furthermore, some strains of resistant bacteria are no longer confined to hospitals and are occurring in otherwise healthy individuals in communities across the United States and other countries. As resistant bacteria multiply, so does the burden they place on our health care system. The economic cost has reached billions of dollars annually in the United States, according to estimates from IOM and the former Congressional Office of Technology Assessment. The human cost in terms of pain, grief, and suffering, however, is incalculable. Lipopeptides are new agents for treating Gram-positive infections November 22nd, Cyclic lipopeptides are new antimicrobials that show promise for the treatment of Gram-positive infections. According to published research from the United States, "The increasing incidence of serious infections caused by antibiotic-resistant Gram-positive bacteria has led to the development of new spectrum-specific agents. One such agent is Cubicin (daptomycin for injection), the first member of a new class of antibacterials called cyclic lipopeptides. Daptomycin has rapid, concentration-dependent bactericidal activity against most clinically significant Gram-positive pathogens, including vancomycin-resistant enterococci, methicillin-resistant Staphylococcus aureus,... 20
 Furthermore, some strains of resistant bacteria are no. longer confined to hospitals and are occurring in otherwise. healthy individuals in communities across the United States. and other countries. As resistant bacteria multiply, so does the burden they. place on our health care system. The economic cost has. reached billions of dollars annually in the United States, according to estimates from IOM and the former. Congressional Office of Technology Assessment. The. human cost in terms of pain, grief, and suffering, however, is incalculable. Lipopeptides are new agents for treating Gram-positive infections. November 22nd, 2004 Cyclic lipopeptides are new antimicrobials that show promise for the treatment of Gram-positive infections. According to published research from the United States, The increasing incidence of serious infections caused by antibiotic-resistant Gram-positive bacteria has led to the development of new spectrum-specific agents. One such agent is Cubicin (daptomycin for injection), the first member of a new class of antibacterials called cyclic lipopeptides. Daptomycin has rapid, concentration-dependent bactericidal activity against most clinically significant Gram-positive pathogens, including vancomycin-resistant enterococci, methicillin-resistant Staphylococcus aureus,")
21
Tigecycline Νο 21

22
Timeline of Staphylococcal antibiotic resistance
Penicillin-resistance Sporadic MRSA Epidemic MRSA GISA CA-MRSA VRSA 1940 1950 1960 1970 1980 1990 2000 2010 22

23
Πολυανθεκτικά παθογόνα με παγκόσμια κατανομή
ESKAPE Enterococcus faecium S. aureus Klebsiella pneumoniae Acinetobacter baumannii Pseudomonas aeruginosa Enterobacter Of the 6 famous ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter species, Pseudomonas aeruginosa, and Enterobacter species) recognized as the most important emerging threats of this century, 4 are gram-negative bacilli (K pneumoniae, Acinetobacter species, P aeruginosa, and Enterobacter species).1 Pseudomonas aeruginosa Acinetobacter baumannii Enterobacteriaceae: Klebsiella pneumoniae Enterobacter spp. (esp cloacae) E. coli Serratia marcescens Clin Infect Dis 2009; 48:1–12 23
 recognized as the most important emerging threats of this century, 4 are gram-negative bacilli (K pneumoniae, Acinetobacter species, P aeruginosa, and Enterobacter species).1. Pseudomonas aeruginosa. Acinetobacter baumannii. Enterobacteriaceae: Klebsiella pneumoniae. Enterobacter spp. (esp cloacae) E. coli. Serratia marcescens. Clin Infect Dis 2009; 48:1–")
25
A Changing Landscape for Numbers of Approved Antibacterial Agents
2 4 6 8 10 12 14 16 18 Number of agents approved Resistance 2008 Bars represent number of new antimicrobial agents approved by the FDA during the period listed. Infectious Diseases Society of America. Bad Bugs, No Drugs. July 2004; Spellberg B et al. Clin Infect Dis. 2004;38: ; New antimicrobial agents. Antimicrob Agents Chemother. 2006;50:1912

27
27

28
Brief history of fight with bacteria disease
2000 B.C. Are you sick? Take this root! Treatment with root is profane. Pray! Pray is superstition. Take this nostrum! Nostrums are charlatanry. Take this pill! Old pills are not efficient. Take penicilin! Oh, bacteria mutated! Take tetracyclin! Other „39 Oh“ Take more effective antibiotic! Bacteria is winning! Take this root!

29
Πολυανθεκτικά Gram αρνητικά παθογόνα (κατά CDC)
Πολυανθεκτικά (Multi-Drug Resistant - MDR): Αντοχή σε ≥3 κατηγορίες αντιβιοτικών* Αυξημένης αντοχής (Extensively drug-resistant XDR): Ευαισθησία σε ≤2 αντιβιοτικά Πανανθεκτικά (Pan resistance): Αντοχή σε όλα τα αντιβιοτικά * Κατηγορίες: β-λακταμικά, κινολόνες, καρβαπενέμες
: Αντοχή σε ≥3 κατηγορίες αντιβιοτικών* Αυξημένης αντοχής (Extensively drug-resistant XDR): Ευαισθησία σε ≤2 αντιβιοτικά. Πανανθεκτικά (Pan resistance): Αντοχή σε όλα τα αντιβιοτικά. * Κατηγορίες: β-λακταμικά, κινολόνες, καρβαπενέμες.")
30
Emergence of MDR Organisms
A new enzyme that will inactivate or destroy the antibiotic New Genetic Material Alteration of the antibiotic target site to evade antibiotic action Bacteria: new proteins Bacteria Prevention of antibiotic access to the target site

32
Vatopoulos A. Euro Surveill 2008;13(4)
")
34
34

35
Methicillin-Resistant Staphylococcus aureus (MRSA), Blood and Spinal Fluid
Source: EARSS, 2008 35

36
E.coli 36

37
Pseudomonas aeruginosa 37

38
WHONET: Αντοχή E. coli σε μη νοσηλευόμενους Ιούλ. – Δεκ. 2005
38

39
ΜΗΧΑΝΙΣΜΟΙ ΑΝΤΟΧΗΣ ΜΙΚΡΟΒΙΩΝ ΣΤΑ ΑΝΤΙΒΙΟΤΙΚΑ
Φυσική αντοχή Επίκτητη αντοχή Αυτόματη μετάλλαξη Μεταβίβαση γενετικού υλικού (πλασμιδιακού ή χρωμοσωμικού) Με σύζευξη (conjugation) Με μεταγωγή με βακτηριοφάγους (transduction) Με μεταμόρφωση (transformation). My son, if they don’t get me, you will become multiresistant
 Με σύζευξη (conjugation) Με μεταγωγή με βακτηριοφάγους (transduction) Με μεταμόρφωση (transformation). My son, if they don’t get me, you will become multiresistant.")
40
Σύζευξη (Conjugation)
Μεταβίβαση DNA (πλασμιδιακού ή επισώματος) από ένα μικροβιακό κύτταρα σε άλλο μέσω συζευτικού ινιδίου (pilus).
 από ένα μικροβιακό κύτταρα σε άλλο μέσω συζευτικού ινιδίου (pilus).")
41
ΜΕΤΑΓΩΓΗ ΓΕΝΕΤΙΚΟΥ ΥΛΙΚΟΥ ΜΙΚΡΟΒΙΩΝ ΜΕ ΦΑΓΟΥΣ (transduction)
")
42
Μεταβίβαση DNA με μεταμόρφωση
(συγγενή μικρόβια)
")
43
Transposon, jumping gene

44
Βιοχημικοί μηχανισμοί αντοχής
Ενζυμική αδρανοποίηση Μεταβολή στόχου -μορίου σύνδεσης και δράσης Μειωμένη διείσδυση στο κύτταρο Αυξημένη αποβολή (ενεργοποίηση αντλιών εκροής) Παράκαμψη μεταβολικής οδού.
 Παράκαμψη μεταβολικής οδού.")
45
Εμφάνιση πολυανθεκτικών (MDR) παθογόνων
Νέο ένζυμο που αδρανοποιεί το αντιβιοτικό Μετάλλαξη Νέο γονίδιο Απόκτηση νέων πρωτεϊνών Μεταβολή στόχου δράσης αντιβιοτικού Βακτήριο Παρεμπόδιση εισόδου ή αποβολή αντιβιοτικού

46
Class B (metallo-b-lactamase)
Καρβαπενεμάσες Class A PC, SME, IMI, NMC, GES, KPC Enterobacteriaceae (σπάνια P. aeruginosa) Class B (metallo-b-lactamase) IMP, VIM, GIM, SPM, NDM-1 P. aeruginosa, Enterobacteriacea, Acinetobacter spp Class D OXA Acinetobacter spp.
 Class B (metallo-b-lactamase) IMP, VIM, GIM, SPM, NDM-1. P. aeruginosa, Enterobacteriacea, Acinetobacter spp. Class D. OXA. Acinetobacter spp.")
47
Β ΛΑΚΤΑΜΑΣΕΣ Ομάδα Είδη Αντοχή* Α Β C D TEM, SHV, CTX-M KPC, GES, SME
Πενικιλλίνες, κεφαλοσ 3ης γεν Όλα τα β λακταμικά Β NDM-1, VIM, IMP, GIM, SPM (μεταλο-β-λακταμάσες) Όλα τα β λακταμικά (?εξαιρείται η αζτρεονάμη;) C AmpC cephamycinases (AmpC) Όλα τα β λακταμικά, εκτός καρβαπενέμες, κεφeπίμη D OXA (OXA-23, -24, -58, -143) Extended-spectrum β- lactamases (ESBLs) Όλα τα β λακταμικά, εκτός καρβαπενέμες, κεφαμυκίνες Most KPC-producing isolates are resistant to fluoroquinolones, aminoglycosides, and co-trimoxazole. Several isolates remain susceptible to amikacin or gentamicin, and most isolates remain susceptible to colistin and tigecycline The blaKPC genes have usually been identified in large plasmids These plasmids usually also carry aminoglycoside-resistance determinants, and have been associated with other β-lactamase genes such as the most widespread ESBL gene, blaCTX-M-15 Up to seven different β lactamases were found associated with blaKPC in one K pneumoniae isolate Enzymes that mediate the destruction of antibiotic molecules contribute more frequently to resistance in gram-negative pathogens. Among the enzymes that warrant particular attention are b-lactamases, which catalyze the hydrolysis of b-lactam antibiotics, including AmpC b-lactamases, metallo-b-lactamases (MBLs), extended-spectrum b-lactamases (ESBLs), oxacillinases, and K. pneumoniae carbapenemases (KPCs) (table 1). Nowhere is the concept of antimicrobial resistance better portrayed than with the gram-negative bacilli, which have proven to be tough adversaries for clinicians and researchers alike. Of the 6 famous ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter species, Pseudomonas aeruginosa, and Enterobacter species) recognized as the most important emerging threats of this century, 4 are gram-negative bacilli (K pneumoniae, Acinetobacter species, P aeruginosa, and Enterobacter species).1 This review will address 3 major types of multidrug-resistant (MDR) gram-negative pathogens: extended-spectrum β-lactamase (ESBL)– producing Enterobacteriaceae, carbapenemase-producing Enterobacteriaceae, and MDR P aeruginosa. The resistance mechanisms exhibited by these organisms and the epidemiology Current Concepts in Antimicrobial Therapy Against Resistant Gram-Negative Organisms: Extended-Spectrum β-Lactamase–Producing Enterobacteriaceae, Carbapenem-Resistant Enterobacteriaceae, and Multidrug-Resistant Pseudomonas aeruginosa Souha S. Kanj, MD, and Zeina A. Kanafani, MD The development of antimicrobial resistance among gram-negative pathogens has been progressive and relentless. Pathogens of particular concern include extended-spectrum β-lactamase– producing Enterobacteriaceae, carbapenem-resistant Enterobacteriaceae, and multidrug-resistant Pseudomonas aeruginosa. Classic agents used to treat these pathogens have become outdated. Of the few new drugs available, many have already become targets for bacterial mechanisms of resistance. This review describes the current approach to infections due to these resistant organisms and elaborates on the available treatment options. Mayo Clin Proc. 2011;86(3): ESBL = extended-spectrum β-lactamase; IV = intravenously; KPC = Klebsiella pneumoniae carbapenemase; MDR = multidrug-resistant; MIC = minimum inhibitory concentration of the infections they cause will be discussed. Existing and emerging therapeutic approaches to each type of organism will then be surveyed. MECHANISMS OF RESISTANCE Production of β-lactamase is the most commonly encountered mechanism of resistance of bacterial pathogens to β-lactam antibiotics. Many enzymes have been described, encoded either by chromosomal genes or by genes located on movable elements such as plasmids and transposons. Classification schemes for β-lactamases are based on molecular structure (Ambler classification)2 or functional similarities (Bush-Jacoby-Medeiros classification)3 (Table 1). Extended-spectrum β-lactamase enzymes initially arose through point mutations in the genes encoding the classic TEM and SHV β-lactamases, thereby generating an array of enzymes with an expanded spectrum of activity.4 The potent hydrolytic activity of CTX-M enzymes against cefotaxime was later recognized. Unlike TEM, SHV, and CTX-M ESBL enzymes that are predominantly expressed by Enterobacteriaceae, the oxacillin-hydrolyzing enzymes have been mostly isolated from P aeruginosa, and some have evolved to exhibit the ESBL phenotype. In contrast to the plasmid-mediated ESBL enzymes, AmpC β-lactamases are predominantly chromosomally encoded.5 Their expression is mostly noted in Enterobacter species, Citrobacter species, and P aeruginosa. Although chromosomal AmpCenzymes are usually poorly expressed in Escherichia coli and Klebsiella species, plasmid-mediated AmpC enzymes can confer β-lactam resistance similar to Enterobacter isolates. Other less commonly encountered ESBL enzymes include PER-1, VEB-1, and BES-1.6 Carbapenemases are the β-lactamases with the widest spectrum of activity. In addition to hydrolyzing carbapenems, carbapenemases are active against most other members of the β-lactam family with few exceptions. The major drive behind the emergence of carbapenemases has been the widespread use of carbapenems both in the empirical and directed treatment of serious infections, which placed selection pressure on bacterial pathogens. On the basis of their molecular structure, carbapenemases belong to the A, B, or D classes of β-lactamase enzymes7 (Table 2). The plasmid-borne K pneumoniae carbapenemases (KPCs) are currently among the most prevalent and widely distributed carbapenemases. They are particularly difficult to detect by microbiology laboratories because many isolates have minimum inhibitory concentrations (MICs) against imipenem or meropenem that, albeit high, remain in the susceptible range.8,9 It has been observed through in vitro studies that ertapenem may be the most appropriate substrate for detection of KPC production.8 Other clinically important carbapenemases include the metallo-β- lactamases and the oxacillin-hydrolyzing carbapenemases. Besides β-lactamase production, P aeruginosa isolates can exhibit additional resistance mechanisms, such as aminoglycoside- modifying enzymes, efflux pumps, porin loss, and various target site modifications.10 class C include the AmpC enzymes; class D include the OXA-type β-lactamases. 1.2 Classification, epidemiology and worldwide spread The most commonly used classification for carbapenemases is that defined by Ambler, although the one by Bush-Jacoby is also used. The Ambler classification separates β-lactamases into four classes A-D, based on their molecular structure [5, 23]. Ambler classes A, B and D will be used throughout this document when referring to carbapenemase classification. An additional classification for carbapenemases has recently been proposed by other experts, whereby extended-spectrum β-lactamases (ESBLs) with hydrolytic activity against carbapenems above a quantitatively defined threshold are designated as ESBLCARBA [24]. Class A carbapenemases are serine β-lactamases and contain serine at their active site [3, 23]. KPC is the most frequently encountered Class A carbapenemase and, along with its variants KPC-2 to KPC-13, which differ solely by amino-acid mutations, it has spread throughout the USA and globally [2]. The bla KPC gene is plasmid-mediated and is transported in a Tn3-based transposon, Tn4401, which makes it readily transferable between bacterial isolates [25]. Following the first report of a K. pneumoniae isolate harbouring blaKPC from USA [1], blaKPC, spread efficiently with patient mobility and disseminated across borders internationally [26-28]. KPCs are predominantly found in Enterobacteriaceae, most commonly in K. pneumoniae isolates, but have recently also been reported in nonfermentative bacteria such as Pseudomonas spp. [29, 30] and Acinetobacter spp. [31]. KPC has now become endemic in many areas of the world, including north-eastern USA [32, 33], Greece [34, 35], Israel [28, 36, 37], Colombia [38] and Puerto Rico [39-40]. Class B carbapenemases, also known as metallo-β-lactamases (MBLs), are zinc-dependent at their active site. Originally, MBLs were described in non-fermentative bacteria such as Pseudomonas spp. and Acinetobacter spp. [41, 42], and more recently have also been described in Enterobacteriaceae [43, 44]. The most commonly found Class B carbapenemases are of the VIM-type, [45] which have been identified on all continents [46], but are found mostly in southern Europe [40, 45, 47]. Evidence for the emergence of newer carbapenemases is the description of a novel type of MBL carbapenemase, the New Delhi metallo-β-lactamase-1 (NDM-1) [48-50], mostly associated with travel to the Indian subcontinent, where it appears to be endemic [49, 51]. NDM-1 has also been reported from other countries including China, Australia [52], the USA [53], Canada [54, 55] and many countries in Europe [49, 50, 56-58], most recently the Balkan region [50, 59-61]. These isolates are reported either as cases of returned travellers from the Indian subcontinent, autochthonous cases in countries with no travel association or contact with infected individuals, or as cases of in-country secondary transmission. A recent report of NDM-2 from the north of Africa is worrying testimony that new variants of NDM have begun to emerge [62]. Over the past decade carbapenemases, a group of clinically important β-lactamases have emerged and spread among Enterobacteriaceae (1-4). One of the milestones in the emergence of carbapenemases in Enterobacteriaceae was the detection of a novel carbapenemase, Klebsiella pneumoniae carbapenemase (KPC), in a Klebsiella pneumoniae isolate in North Carolina, USA, which later successfully spread throughout the world [1]. Since then, most acquired carbapenemases have been found and reported in carbapenemase-producing Enterobacteriaceae (CPE) globally [2, 3]. Carbapenemases are enzymes that can efficiently hydrolyse most β-lactams, including carbapenems [4, 5]. In addition, many CPE strains frequently carry additional resistance determinants to other non-β-lactam antibiotics, making these organisms resistant to most antibiotics. CPE commonly remain susceptible to only a few classes of antimicrobials, commonly the polymyxins, tigecycline, fosfomycin, and nitrofurantoin. There is no proven clinical efficacy against these strains and in fact there are reports of clinical failures [6] and emerging resistance to these antimicrobials [7-10]. The emergence and spread of CPE has also been identified as a public health threat, especially since recent studies on CPE [11, 12] and carbapenem-non-susceptible Enterobacteriaceae (CNSE) [13, 14] have shown that infection or colonisation has been associated with higher in-hospital mortality. Similarly, prior studies of outcomes, involving patients infected with multidrug-resistant organisms (MDROs), show that an inadequate choice or the delayed administration of antimicrobial therapy is associated with poorer patient outcomes, increased morbidity, mortality, increased length of hospital stay and increased costs [15-20]. The risk to patients infected with these MDROs becomes even greater, given the very limited number of new antimicrobial agents that are in the developmental pipeline [21, 22]. Class D carbapenemases are oxacillinases [46] and include the OXA-type carbapenemases, predominantly in Acinetobacter spp. (mainly OXA-23, -24, -58, and -143) [46, 63, 64] but also in P. aeruginosa (mainly OXA-40) [65]. The first report of OXA-48 in Enterobacteriaceae was from Turkey [66, 67] and it has since been reported from other countries in the Mediterranean basin, including Israel [68], Tunisia [69], Morocco [70] and Spain [71]. Cross-border transfer of OXA-48-producing Enterobacteriaceae into healthcare facilities in the destination countries (e.g. France which has seen a sharp increase in cases recently) is being reported more frequently in the literature, suggesting that the mode of introduction into healthcare facilities is patient mobility [72-74]. with metallo-β-lactamases (IMP, NDM or VIM) and non-metallo (KPC and OXA-48) enzymes as well as those combining an extended-spectrum β-lactamase (ESBL) or AmpC enzyme with porin loss. Συνήθως συνυπάρχει αντοχή σε φθοριοκινολόνες, αμινογλυκοσίδες και TMP-SMX (γονίδια αντοχής στα ίδια πλασμίδια) Kanj SS, Kanafani ZA. Mayo Clin Proc. 2011;86:250-9.
 Όλα τα β λακταμικά ( εξαιρείται η αζτρεονάμη;) C. AmpC cephamycinases (AmpC) Όλα τα β λακταμικά, εκτός καρβαπενέμες, κεφeπίμη. D. OXA (OXA-23, -24, -58, -143) Extended-spectrum β- lactamases (ESBLs) Όλα τα β λακταμικά, εκτός καρβαπενέμες, κεφαμυκίνες. Most KPC-producing isolates are resistant to fluoroquinolones, aminoglycosides, and co-trimoxazole. Several isolates remain susceptible to amikacin or gentamicin, and most isolates remain susceptible to colistin and tigecycline. The blaKPC genes have usually been identified in large plasmids. These plasmids usually also carry aminoglycoside-resistance determinants, and have been associated with other β-lactamase genes such as the most widespread ESBL gene, blaCTX-M-15. Up to seven different β lactamases were found associated with blaKPC in one K pneumoniae isolate. Enzymes that mediate the destruction of antibiotic molecules contribute more frequently to resistance in gram-negative pathogens. Among the enzymes that warrant particular attention are b-lactamases, which catalyze the hydrolysis of b-lactam antibiotics, including AmpC b-lactamases, metallo-b-lactamases (MBLs), extended-spectrum b-lactamases (ESBLs), oxacillinases, and K. pneumoniae carbapenemases (KPCs) (table 1). Nowhere is the concept of antimicrobial resistance better. portrayed than with the gram-negative bacilli, which have. proven to be tough adversaries for clinicians and researchers. alike. Of the 6 famous ESKAPE pathogens (Enterococcus. faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter species, Pseudomonas aeruginosa, and. Enterobacter species) recognized as the most important. emerging threats of this century, 4 are gram-negative bacilli. (K pneumoniae, Acinetobacter species, P aeruginosa, and. Enterobacter species).1 This review will address 3 major. types of multidrug-resistant (MDR) gram-negative pathogens: extended-spectrum β-lactamase (ESBL)– producing. Enterobacteriaceae, carbapenemase-producing Enterobacteriaceae, and MDR P aeruginosa. The resistance mechanisms. exhibited by these organisms and the epidemiology. Current Concepts in Antimicrobial Therapy Against Resistant. Gram-Negative Organisms: Extended-Spectrum β-Lactamase–Producing. Enterobacteriaceae, Carbapenem-Resistant Enterobacteriaceae, and. Multidrug-Resistant Pseudomonas aeruginosa. Souha S. Kanj, MD, and Zeina A. Kanafani, MD. The development of antimicrobial resistance among gram-negative. pathogens has been progressive and relentless. Pathogens. of particular concern include extended-spectrum β-lactamase– producing Enterobacteriaceae, carbapenem-resistant Enterobacteriaceae, and multidrug-resistant Pseudomonas aeruginosa. Classic agents used to treat these pathogens have become outdated. Of the few new drugs available, many have already become. targets for bacterial mechanisms of resistance. This review describes. the current approach to infections due to these resistant. organisms and elaborates on the available treatment options. Mayo Clin Proc. 2011;86(3): ESBL = extended-spectrum β-lactamase; IV = intravenously; KPC = Klebsiella pneumoniae carbapenemase; MDR = multidrug-resistant; MIC = minimum inhibitory concentration. of the infections they cause will be discussed. Existing and. emerging therapeutic approaches to each type of organism. will then be surveyed. MECHANISMS OF RESISTANCE. Production of β-lactamase is the most commonly encountered. mechanism of resistance of bacterial pathogens to. β-lactam antibiotics. Many enzymes have been described, encoded either by chromosomal genes or by genes located. on movable elements such as plasmids and transposons. Classification schemes for β-lactamases are based on molecular. structure (Ambler classification)2 or functional similarities. (Bush-Jacoby-Medeiros classification)3 (Table 1). Extended-spectrum β-lactamase enzymes initially arose. through point mutations in the genes encoding the classic. TEM and SHV β-lactamases, thereby generating an. array of enzymes with an expanded spectrum of activity.4. The potent hydrolytic activity of CTX-M enzymes against. cefotaxime was later recognized. Unlike TEM, SHV, and. CTX-M ESBL enzymes that are predominantly expressed. by Enterobacteriaceae, the oxacillin-hydrolyzing enzymes. have been mostly isolated from P aeruginosa, and some. have evolved to exhibit the ESBL phenotype. In contrast to. the plasmid-mediated ESBL enzymes, AmpC β-lactamases. are predominantly chromosomally encoded.5 Their expression. is mostly noted in Enterobacter species, Citrobacter. species, and P aeruginosa. Although chromosomal AmpCenzymes are usually poorly expressed in Escherichia coli. and Klebsiella species, plasmid-mediated AmpC enzymes. can confer β-lactam resistance similar to Enterobacter. isolates. Other less commonly encountered ESBL enzymes. include PER-1, VEB-1, and BES-1.6. Carbapenemases are the β-lactamases with the widest. spectrum of activity. In addition to hydrolyzing carbapenems, carbapenemases are active against most other. members of the β-lactam family with few exceptions. The. major drive behind the emergence of carbapenemases has. been the widespread use of carbapenems both in the empirical. and directed treatment of serious infections, which. placed selection pressure on bacterial pathogens. On the. basis of their molecular structure, carbapenemases belong. to the A, B, or D classes of β-lactamase enzymes7 (Table. 2). The plasmid-borne K pneumoniae carbapenemases. (KPCs) are currently among the most prevalent and widely. distributed carbapenemases. They are particularly difficult. to detect by microbiology laboratories because many. isolates have minimum inhibitory concentrations (MICs) against imipenem or meropenem that, albeit high, remain. in the susceptible range.8,9 It has been observed through in. vitro studies that ertapenem may be the most appropriate. substrate for detection of KPC production.8 Other clinically. important carbapenemases include the metallo-β- lactamases and the oxacillin-hydrolyzing carbapenemases. Besides β-lactamase production, P aeruginosa isolates can. exhibit additional resistance mechanisms, such as aminoglycoside- modifying enzymes, efflux pumps, porin loss, and various target site modifications.10. class C include the AmpC enzymes; class D include the OXA-type β-lactamases. 1.2 Classification, epidemiology and worldwide spread. The most commonly used classification for carbapenemases is that defined by Ambler, although the one by Bush-Jacoby is also used. The Ambler classification separates β-lactamases into four classes A-D, based on their molecular structure [5, 23]. Ambler classes A, B and D will be used throughout this document when referring to carbapenemase classification. An additional classification for carbapenemases has recently been proposed by other experts, whereby extended-spectrum β-lactamases (ESBLs) with hydrolytic activity against carbapenems. above a quantitatively defined threshold are designated as ESBLCARBA [24]. Class A carbapenemases are serine β-lactamases and contain serine at their active site [3, 23]. KPC is the most frequently encountered Class A carbapenemase and, along with its variants KPC-2 to KPC-13, which differ solely by amino-acid mutations, it has spread throughout the USA and globally [2]. The bla KPC gene is plasmid-mediated and is transported in a Tn3-based transposon, Tn4401, which makes it readily transferable between bacterial isolates [25]. Following the first report of a K. pneumoniae isolate harbouring blaKPC from USA [1], blaKPC, spread efficiently with patient mobility and disseminated across borders internationally [26-28]. KPCs are predominantly found in Enterobacteriaceae, most commonly in K. pneumoniae isolates, but have recently also been reported in nonfermentative bacteria such as Pseudomonas spp. [29, 30] and Acinetobacter spp. [31]. KPC has now become endemic in many areas of the world, including north-eastern USA [32, 33], Greece [34, 35], Israel [28, 36, 37], Colombia [38] and Puerto Rico [39-40]. Class B carbapenemases, also known as metallo-β-lactamases (MBLs), are zinc-dependent at their active site. Originally, MBLs were described in non-fermentative bacteria such as Pseudomonas spp. and Acinetobacter spp. [41, 42], and more recently have also been described in Enterobacteriaceae [43, 44]. The most commonly found. Class B carbapenemases are of the VIM-type, [45] which have been identified on all continents [46], but are. found mostly in southern Europe [40, 45, 47]. Evidence for the emergence of newer carbapenemases is the. description of a novel type of MBL carbapenemase, the New Delhi metallo-β-lactamase-1 (NDM-1) [48-50], mostly associated with travel to the Indian subcontinent, where it appears to be endemic [49, 51]. NDM-1 has. also been reported from other countries including China, Australia [52], the USA [53], Canada [54, 55] and many. countries in Europe [49, 50, 56-58], most recently the Balkan region [50, 59-61]. These isolates are reported. either as cases of returned travellers from the Indian subcontinent, autochthonous cases in countries with no. travel association or contact with infected individuals, or as cases of in-country secondary transmission. A recent. report of NDM-2 from the north of Africa is worrying testimony that new variants of NDM have begun to emerge. [62]. Over the past decade carbapenemases, a group of clinically important β-lactamases have emerged and spread. among Enterobacteriaceae (1-4). One of the milestones in the emergence of carbapenemases in. Enterobacteriaceae was the detection of a novel carbapenemase, Klebsiella pneumoniae carbapenemase (KPC), in a Klebsiella pneumoniae isolate in North Carolina, USA, which later successfully spread throughout the world. [1]. Since then, most acquired carbapenemases have been found and reported in carbapenemase-producing. Enterobacteriaceae (CPE) globally [2, 3]. Carbapenemases are enzymes that can efficiently hydrolyse most β-lactams, including carbapenems [4, 5]. In. addition, many CPE strains frequently carry additional resistance determinants to other non-β-lactam antibiotics, making these organisms resistant to most antibiotics. CPE commonly remain susceptible to only a few classes of. antimicrobials, commonly the polymyxins, tigecycline, fosfomycin, and nitrofurantoin. There is no proven clinical. efficacy against these strains and in fact there are reports of clinical failures [6] and emerging resistance to these. antimicrobials [7-10]. The emergence and spread of CPE has also been identified as a public health threat, especially since recent. studies on CPE [11, 12] and carbapenem-non-susceptible Enterobacteriaceae (CNSE) [13, 14] have shown that. infection or colonisation has been associated with higher in-hospital mortality. Similarly, prior studies of. outcomes, involving patients infected with multidrug-resistant organisms (MDROs), show that an inadequate. choice or the delayed administration of antimicrobial therapy is associated with poorer patient outcomes, increased morbidity, mortality, increased length of hospital stay and increased costs [15-20]. The risk to patients. infected with these MDROs becomes even greater, given the very limited number of new antimicrobial agents. that are in the developmental pipeline [21, 22]. Class D carbapenemases are oxacillinases [46] and include the OXA-type carbapenemases, predominantly in. Acinetobacter spp. (mainly OXA-23, -24, -58, and -143) [46, 63, 64] but also in P. aeruginosa (mainly OXA-40) [65]. The first report of OXA-48 in Enterobacteriaceae was from Turkey [66, 67] and it has since been reported. from other countries in the Mediterranean basin, including Israel [68], Tunisia [69], Morocco [70] and Spain [71]. Cross-border transfer of OXA-48-producing Enterobacteriaceae into healthcare facilities in the destination. countries (e.g. France which has seen a sharp increase in cases recently) is being reported more frequently in. the literature, suggesting that the mode of introduction into healthcare facilities is patient mobility [72-74]. with metallo-β-lactamases (IMP, NDM or VIM) and non-metallo (KPC and OXA-48) enzymes as well as those combining an extended-spectrum β-lactamase (ESBL) or AmpC enzyme with porin loss. Συνήθως συνυπάρχει αντοχή σε φθοριοκινολόνες, αμινογλυκοσίδες και TMP-SMX (γονίδια αντοχής στα ίδια πλασμίδια) Kanj SS, Kanafani ZA. Mayo Clin Proc. 2011;86:")
49
NDM-1 — A Cause for Worldwide Concern
Robert C. Moellering, Jr., M.D. N Engl J Med 2010; 363: Welcome Guest Renew, Subscribe or Create Account Sign In The New England Journal of Medicine Home Articles Issues Specialties & Topics For Authors Advanced Search Perspective NDM-1 — A Cause for Worldwide Concern Robert C. Moellering, Jr., M.D. N Engl J Med 2010; 363: December 16, 2010 Article References The past several years have seen a number of reports of superbugs: methicillin-resistant Staphylococcus aureus, the so-called ESKAPE organisms (an acronym for Enterococcus faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and enterobacter species), and others.1 For the most part, these organisms owe their superbug status not to enhanced pathogenicity or virulence (although some are capable of causing overwhelming disease in the proper setting) but to their resistance to multiple antimicrobial agents. The most recent reports of superbugs in the professional and lay literature discuss NDM-1, which stands for New Delhi metallo-beta-lactamase 1 and actually refers not to a single bacterial species but to a transmissible genetic element encoding multiple resistance genes that was initially isolated from a strain of klebsiella obtained from a patient who acquired the organism in New Delhi, India.2 Subsequently, organisms in the Enterobacteriaceae family containing this genetic element (or variants thereof) have been found widely throughout India, Pakistan, and Bangladesh and are now turning up in Britain and, in rapid order, many other countries around the world. The spread of these organisms has prompted widespread concern because some of them are resistant to all antimicrobial agents except the polymyxins. Concern about antimicrobial resistance in bacteria is not new, however. This fact is clearly reflected in articles published 50 years ago in the Journal. A 1960 editorial accompanying an article on novobiocin and tetracycline decried the overuse of antibiotics and the irrational use of fixed combinations of antimicrobials, which were widely manufactured and prescribed by the pharmaceutical industry at that time. Another article on the transmissibility of staphylococci noted that the administration of tetracycline to hospitalized patients increased the rate of nasopharyngeal colonization with S. aureus, much of which showed resistance to tetracycline. Another Journal editorial on antibiotic resistance quoted a study from Hammersmith Hospital clearly showing that limiting the use of antimicrobial agents in the hospital setting was associated with a decrease in resistance to penicillin and tetracycline among staphylococci. Thus, as of 50 years ago, most of the important principles concerning the nature, dissemination, and potential control of antibiotic resistance were known: the role of inappropriate antibiotic use in selecting resistant organisms, the ability of resistant organisms to spread in the hospital setting, and the value of limiting antibiotic use in the hospital as a control measure. Despite such knowledge and vigorous worldwide attempts to initiate methods to prevent and control antibiotic resistance, our success has been limited at best. The story of the beta-lactamases is a case in point. The first enzyme capable of destroying penicillin was described before the initial clinical application of penicillin in the early 1940s. The discovery of compounds resistant to beta-lactamases (e.g, cephalosporins and carbapenems) or capable of inactivating them (e.g., beta-lactamase inhibitors) has simply been met with the evolution of new beta-lactamases, often through mutations that inactivate these antibiotics. At present, more than 890 such unique enzymes have been discovered — far more than the antibiotics developed to combat them.3 Some of these enzymes are chromosomally mediated, but the majority of them are found on transmissible genetic elements, and for the most part, acquisition of the resistance genes does not result in a huge fitness cost to the recipient organism. This is not true of all types of resistance, however. Resistance to oxazolidinones, such as linezolid in the case of S. aureus, is an example of a resistance mechanism that does extract a fitness cost. Because the oxazolidinone target on the 23S ribosomal RNA of the ribosome exists in multiple (four to six) copies, it requires several mutations for organisms to develop resistance, and these mutations are clearly associated with a decrease in fitness. This may account for the fact that for the first decade or so of linezolid use, the emergence of resistance in S. aureus has been relatively uncommon. However, nature clearly abhors a vacuum, and recently a new mechanism of resistance has been discovered, initially in staphylococci in swine in Germany. The mechanism of resistance in these organisms turned out to be attributable to genes that encoded a methylase that altered the ribosomal binding sites for linezolid and caused cross-resistance to a number of other ribosomally active antimicrobials, including chloramphenicol, clindamycin, the pleuromutilins, and streptogramin B — a case of a single enzyme knocking out the activity of five different classes of antibiotics. Worse yet, the gene encoding this methylase is found on a transmissible plasmid, and recent outbreaks of linezolid resistance resulting from this mechanism in Madrid suggest that the era of almost universal susceptibility of S. aureus to linezolid may be coming to an end.4 All of which brings us back to NDM-1. What makes this enzyme so frightening is not only its intrinsic ability to destroy most known beta-lactam antibiotics but also the company it keeps. As noted earlier, this enzyme was initially discovered in a strain of K. pneumoniae from a Swedish patient of Indian origin who traveled to New Delhi and acquired a urinary tract infection there. The original organism was found to be resistant to all antimicrobial agents tested except colistin.5 Molecular examination of the isolate revealed that it contained a novel metallo-beta-lactamase that readily hydrolyzed penicillins, cephalosporins, and carbapenems (with the exception of aztreonam). The gene encoding this novel beta-lactamase (which had not been known previously) was found on a large 180-kb resistance-conferring genetic element that was easily transferred to other Enterobacteriaceae and that contained a variety of other resistance determinants, including a gene encoding another broad-spectrum beta-lactamase (CMY-4) and genes inactivating erythromycin, ciprofloxacin, rifampicin, and chloramphenicol. In addition, the genetic element encoded an efflux pump capable of causing additional antimicrobial resistance and growth promoters that insured the transcription of the genes contained in the genetic element.2 The Origin and Spread of NDM-1. K. pneumoniae containing NDM-1 was first discovered in By 2009, a study in Mumbai revealed 24 carbapenem-resistant Enterobacteriaceae, 22 of which were NDM-1 producers. Of these 22 organisms, 10 were klebsiella species, 9 were Escherichia coli, 2 were enterobacter species, and 1 was Morganella morganii — illustrating the ability of the plasmid to spread rapidly among strains of Enterobacteriaceae. A more extensive recent study has shown widespread distribution of NDM-1–producing Enterobacteriaceae in Bangladesh, India, Pakistan, and Britain, all of which appears to have occurred since the original isolate was discovered in In addition, isolates of Enterobacteriaceae-containing NDM-1 have now been characterized in the United States, Israel, Turkey, China, India, Australia, France, Japan, Kenya, Singapore, Taiwan, and the Nordic countries. The enzyme has been isolated from three different bacterial species in the United States, including klebsiella, E. coli, and enterobacter. Thus far, the majority of isolates in countries throughout the world can be traced to subjects who have traveled to India to visit family or have received medical care there. However, the ability of this genetic element to spread rapidly among Enterobacteriaceae means that there will almost certainly be numerous secondary cases throughout the world that are unrelated to travel to the Indian subcontinent. The conditions leading to the emergence of NDM-1 on the Indian subcontinent will probably never be known with absolute certainty, but the fact that there is widespread nonprescription use of antibiotics in India, a country in which some areas have less than ideal sanitation and a high prevalence of diarrheal disease and crowding, sets the ideal stage for the development of such resistance.2 As frightening as the prospects of the widespread dissemination of NDM-1 are, it is not the only worldwide threat posed by antibiotic resistance. In the United States alone, we have had major outbreaks of infections due to multiresistant klebsiella containing so-called KPC enzymes, which are capable of hydrolyzing the carbapenems and other beta-lactam antibiotics. CTX beta-lactamases are now being found in increasing numbers in isolates of Enterobacteriaceae obtained from outpatients throughout the world and, at the very least, will compromise our ability to use beta-lactam antibiotics to treat community-acquired urinary tract infections. Thus, bacteria continue to thwart our best efforts to contain them and destroy them with antibiotics. How do they do it? They overwhelm us with their superior numbers, they reproduce with remarkable speed, and they develop extremely efficient ways to exchange and promulgate resistance genes. As one of my colleagues, Dr. Adolf Karchmer, put it, “If you reproduced every 20 minutes, you would get smart quickly, too.” In the case of NDM-1, the Centers for Disease Control and Prevention assures us that it can be contained with standard infection-control methods, but history warns us that this is not likely to be the final answer, even in developed countries. Early experience with NDM-1 has shown that it has all the properties necessary to turn organisms that contain it into superbugs after all. Relevant Articles from the NEJM Archives. Antibiotics in fixed combination. N Engl J Med 1960;262:255-6. Hirsch HA, Finland M, Wilcox C, Yarrows J. Antibiotic combinations — antibacterial action of serum after ingestion of novobiocin or tetracycline or both. N Engl J Med 1960;262: Berntsen CA, McDermott W. Increased transmissibility of staphylococci to patients receiving an antimicrobial drug. N Engl J Med 1960;262: Reversing the tide of antibiotic resistance. N Engl J Med 1960;262:578-9. Disclosure forms provided by the author are available with the full text of this article at NEJM.org. Source Information From Harvard Medical School and Beth Israel Deaconess Medical Center — both in Boston. Tools PDF Print Download Citation Save Article Alert Submit a Letter Reprints & Permissions Topics Bacterial Infections Global Health Genetics General More In December 16, 2010 Trends: Most Viewed (LAST WEEK) GENOMICS, TYPE 2 DIABETES, AND OBESITY DECEMBER 9, 2010 THE ORIGIN OF THE HAITIAN CHOLERA OUTBREAK STRAIN LONG-TERM CONTROL OF HIV BY CCR5 DELTA32/DELTA32 STEM-CELL TRANSPLANTATION FEBRUARY 12, 2009 More Trends Content: Current Issue Issue Index Multimedia & Images Archive Information For: Authors Reviewers Subscribers Institutions Media Advertisers Services: Subscribe Renew Pay Bill Activate Subscription Create or Manage Account Alerts RSS & Podcasts Submit a Manuscript Help Resources: Physician Jobs Weekly CME Program Review CME Program Medical Meetings Conventions FAQs Journal Watch NEJM: About Product Information Editors & Publishers Terms of Use Privacy Policy Copyright Advertising Policies Contact Us Follow us Manage Account Manage Alerts Manage Saved Items Article Category Research Reviews Clinical Cases Commentary Other Browse all articles Multimedia Videos in Clinical Medicine Images in Clinical Medicine Interactive Medical Cases Weekly Audio Summaries Browse all multimedia This Week Last Week Browse full index Selected Specialties Cardiology Endocrinology Gastroenterology Genetics Hematology/Oncology Infectious Disease Nephrology Neurology/Neurosurgery Obstetrics/Gynecology Pediatrics Pulmonary/Critical Care Surgery Browse all Specialties & Topics Featured Topics Health Policy and Reform Haiti after the Earthquake Author Center Submit a Manuscript or Letter Track a Manuscript NEJM.org Copyright © 2010 Massachusetts Medical Society. All rights reserved.
, and others.1 For the most part, these organisms owe their superbug status not to enhanced pathogenicity or virulence (although some are capable of causing overwhelming disease in the proper setting) but to their resistance to multiple antimicrobial agents. The most recent reports of superbugs in the professional and lay literature discuss NDM-1, which stands for New Delhi metallo-beta-lactamase 1 and actually refers not to a single bacterial species but to a transmissible genetic element encoding multiple resistance genes that was initially isolated from a strain of klebsiella obtained from a patient who acquired the organism in New Delhi, India.2 Subsequently, organisms in the Enterobacteriaceae family containing this genetic element (or variants thereof) have been found widely throughout India, Pakistan, and Bangladesh and are now turning up in Britain and, in rapid order, many other countries around the world. The spread of these organisms has prompted widespread concern because some of them are resistant to all antimicrobial agents except the polymyxins. Concern about antimicrobial resistance in bacteria is not new, however. This fact is clearly reflected in articles published 50 years ago in the Journal. A 1960 editorial accompanying an article on novobiocin and tetracycline decried the overuse of antibiotics and the irrational use of fixed combinations of antimicrobials, which were widely manufactured and prescribed by the pharmaceutical industry at that time. Another article on the transmissibility of staphylococci noted that the administration of tetracycline to hospitalized patients increased the rate of nasopharyngeal colonization with S. aureus, much of which showed resistance to tetracycline. Another Journal editorial on antibiotic resistance quoted a study from Hammersmith Hospital clearly showing that limiting the use of antimicrobial agents in the hospital setting was associated with a decrease in resistance to penicillin and tetracycline among staphylococci. Thus, as of 50 years ago, most of the important principles concerning the nature, dissemination, and potential control of antibiotic resistance were known: the role of inappropriate antibiotic use in selecting resistant organisms, the ability of resistant organisms to spread in the hospital setting, and the value of limiting antibiotic use in the hospital as a control measure. Despite such knowledge and vigorous worldwide attempts to initiate methods to prevent and control antibiotic resistance, our success has been limited at best. The story of the beta-lactamases is a case in point. The first enzyme capable of destroying penicillin was described before the initial clinical application of penicillin in the early 1940s. The discovery of compounds resistant to beta-lactamases (e.g, cephalosporins and carbapenems) or capable of inactivating them (e.g., beta-lactamase inhibitors) has simply been met with the evolution of new beta-lactamases, often through mutations that inactivate these antibiotics. At present, more than 890 such unique enzymes have been discovered — far more than the antibiotics developed to combat them.3. Some of these enzymes are chromosomally mediated, but the majority of them are found on transmissible genetic elements, and for the most part, acquisition of the resistance genes does not result in a huge fitness cost to the recipient organism. This is not true of all types of resistance, however. Resistance to oxazolidinones, such as linezolid in the case of S. aureus, is an example of a resistance mechanism that does extract a fitness cost. Because the oxazolidinone target on the 23S ribosomal RNA of the ribosome exists in multiple (four to six) copies, it requires several mutations for organisms to develop resistance, and these mutations are clearly associated with a decrease in fitness. This may account for the fact that for the first decade or so of linezolid use, the emergence of resistance in S. aureus has been relatively uncommon. However, nature clearly abhors a vacuum, and recently a new mechanism of resistance has been discovered, initially in staphylococci in swine in Germany. The mechanism of resistance in these organisms turned out to be attributable to genes that encoded a methylase that altered the ribosomal binding sites for linezolid and caused cross-resistance to a number of other ribosomally active antimicrobials, including chloramphenicol, clindamycin, the pleuromutilins, and streptogramin B — a case of a single enzyme knocking out the activity of five different classes of antibiotics. Worse yet, the gene encoding this methylase is found on a transmissible plasmid, and recent outbreaks of linezolid resistance resulting from this mechanism in Madrid suggest that the era of almost universal susceptibility of S. aureus to linezolid may be coming to an end.4. All of which brings us back to NDM-1. What makes this enzyme so frightening is not only its intrinsic ability to destroy most known beta-lactam antibiotics but also the company it keeps. As noted earlier, this enzyme was initially discovered in a strain of K. pneumoniae from a Swedish patient of Indian origin who traveled to New Delhi and acquired a urinary tract infection there. The original organism was found to be resistant to all antimicrobial agents tested except colistin.5 Molecular examination of the isolate revealed that it contained a novel metallo-beta-lactamase that readily hydrolyzed penicillins, cephalosporins, and carbapenems (with the exception of aztreonam). The gene encoding this novel beta-lactamase (which had not been known previously) was found on a large 180-kb resistance-conferring genetic element that was easily transferred to other Enterobacteriaceae and that contained a variety of other resistance determinants, including a gene encoding another broad-spectrum beta-lactamase (CMY-4) and genes inactivating erythromycin, ciprofloxacin, rifampicin, and chloramphenicol. In addition, the genetic element encoded an efflux pump capable of causing additional antimicrobial resistance and growth promoters that insured the transcription of the genes contained in the genetic element.2. The Origin and Spread of NDM-1. K. pneumoniae containing NDM-1 was first discovered in By 2009, a study in Mumbai revealed 24 carbapenem-resistant Enterobacteriaceae, 22 of which were NDM-1 producers. Of these 22 organisms, 10 were klebsiella species, 9 were Escherichia coli, 2 were enterobacter species, and 1 was Morganella morganii — illustrating the ability of the plasmid to spread rapidly among strains of Enterobacteriaceae. A more extensive recent study has shown widespread distribution of NDM-1–producing Enterobacteriaceae in Bangladesh, India, Pakistan, and Britain, all of which appears to have occurred since the original isolate was discovered in In addition, isolates of Enterobacteriaceae-containing NDM-1 have now been characterized in the United States, Israel, Turkey, China, India, Australia, France, Japan, Kenya, Singapore, Taiwan, and the Nordic countries. The enzyme has been isolated from three different bacterial species in the United States, including klebsiella, E. coli, and enterobacter. Thus far, the majority of isolates in countries throughout the world can be traced to subjects who have traveled to India to visit family or have received medical care there. However, the ability of this genetic element to spread rapidly among Enterobacteriaceae means that there will almost certainly be numerous secondary cases throughout the world that are unrelated to travel to the Indian subcontinent. The conditions leading to the emergence of NDM-1 on the Indian subcontinent will probably never be known with absolute certainty, but the fact that there is widespread nonprescription use of antibiotics in India, a country in which some areas have less than ideal sanitation and a high prevalence of diarrheal disease and crowding, sets the ideal stage for the development of such resistance.2. As frightening as the prospects of the widespread dissemination of NDM-1 are, it is not the only worldwide threat posed by antibiotic resistance. In the United States alone, we have had major outbreaks of infections due to multiresistant klebsiella containing so-called KPC enzymes, which are capable of hydrolyzing the carbapenems and other beta-lactam antibiotics. CTX beta-lactamases are now being found in increasing numbers in isolates of Enterobacteriaceae obtained from outpatients throughout the world and, at the very least, will compromise our ability to use beta-lactam antibiotics to treat community-acquired urinary tract infections. Thus, bacteria continue to thwart our best efforts to contain them and destroy them with antibiotics. How do they do it They overwhelm us with their superior numbers, they reproduce with remarkable speed, and they develop extremely efficient ways to exchange and promulgate resistance genes. As one of my colleagues, Dr. Adolf Karchmer, put it, If you reproduced every 20 minutes, you would get smart quickly, too. In the case of NDM-1, the Centers for Disease Control and Prevention assures us that it can be contained with standard infection-control methods, but history warns us that this is not likely to be the final answer, even in developed countries. Early experience with NDM-1 has shown that it has all the properties necessary to turn organisms that contain it into superbugs after all. Relevant Articles from the NEJM Archives. Antibiotics in fixed combination. N Engl J Med 1960;262: Hirsch HA, Finland M, Wilcox C, Yarrows J. Antibiotic combinations — antibacterial action of serum after ingestion of novobiocin or tetracycline or both. N Engl J Med 1960;262: Berntsen CA, McDermott W. Increased transmissibility of staphylococci to patients receiving an antimicrobial drug. N Engl J Med 1960;262: Reversing the tide of antibiotic resistance. N Engl J Med 1960;262: Disclosure forms provided by the author are available with the full text of this article at NEJM.org. Source Information. From Harvard Medical School and Beth Israel Deaconess Medical Center — both in Boston. Tools. PDF. Print. Download Citation. . Save. Article Alert. Submit a Letter. Reprints & Permissions. Topics. Bacterial Infections. Global Health. Genetics General. More In. December 16, Trends: Most Viewed (LAST WEEK) GENOMICS, TYPE 2 DIABETES, AND OBESITY. DECEMBER 9, THE ORIGIN OF THE HAITIAN CHOLERA OUTBREAK STRAIN. LONG-TERM CONTROL OF HIV BY CCR5 DELTA32/DELTA32 STEM-CELL TRANSPLANTATION. FEBRUARY 12, More Trends. Content: Current Issue. Issue Index. Multimedia & Images. Archive Information For: Authors. Reviewers. Subscribers. Institutions. Media. Advertisers. Services: Subscribe. Renew. Pay Bill. Activate Subscription. Create or Manage Account. Alerts. RSS & Podcasts. Submit a Manuscript. Help. Resources: Physician Jobs. Weekly CME Program. Review CME Program. Medical Meetings. Conventions. FAQs. Journal Watch. NEJM: About. Product Information. Editors & Publishers. Terms of Use. Privacy Policy. Copyright. Advertising Policies. Contact Us. Follow us. Manage Account. Manage Alerts. Manage Saved Items. Article Category. Research. Reviews. Clinical Cases. Commentary. Other. Browse all articles. Multimedia. Videos in Clinical Medicine. Images in Clinical Medicine. Interactive Medical Cases. Weekly Audio Summaries. Browse all multimedia. This Week. Last Week. Browse full index. Selected Specialties. Cardiology. Endocrinology. Gastroenterology. Genetics. Hematology/Oncology. Infectious Disease. Nephrology. Neurology/Neurosurgery. Obstetrics/Gynecology. Pediatrics. Pulmonary/Critical Care. Surgery. Browse all Specialties & Topics. Featured Topics. Health Policy and Reform. Haiti after the Earthquake. Author Center. Submit a Manuscript or Letter. Track a Manuscript. NEJM.org Copyright © 2010 Massachusetts Medical Society. All rights reserved.")
50
50

51
Selection for antimicrobial-resistant Strains
Campaign to Prevent Antimicrobial Resistance in Healthcare Settings Selection for antimicrobial-resistant Strains x Resistant Strains Rare x x Antimicrobial Exposure x x x Once resistant strains of bacteria are present in a population, exposure to antimicrobial drugs favors their survival. Reducing antimicrobial selection pressure is one key to preventing antimicrobial resistance and preserving the utility of available drugs for as long as possible. Resistant Strains Dominant 51 51

52
ΚΑΤΑΝΑΛΩΣΗ ΑΝΤΙΒΙΟΤΙΚΩΝ ΚΑΙ ΜΙΚΡΟΒΙΑΚΗ ΑΝΤΟΧΗ
Implementation of control programme Carbapenem-resistant Pseudomonas aeruginosa (%) Carbapenem use (DDDs) 52
 Carbapenem use (DDDs) 52.")
53
ANTIMETΩΠΙΣΗ ΑΝΘΕΚΤΙΚΩΝ ΣΤΙΣ ΚΑΡΒΑΠΕΝΕΜΕΣ ΠΑΘΟΓΟΝΩΝ ΣΕ ΜΕΘ

55
ΣΤΟΧΟΣ ΑΝΤΙΜΙΚΡΟΒΙΑΚΗΣ ΘΕΡΑΠΕΙΑΣ
Η επιτυχής θεραπεία της λοίμωξης Η ελαχιστοποίηση ανάπτυξης αντοχής

56
ΚΥΡΙΟΙ ΤΡΟΠΟΙ ΠΡΟΛΗΨΗΣ & ΕΛΕΓΧΟΥ ΑΝΤΙΜΙΚΡΟΒΙΑΚΗΣ ΑΝΤΟΧΗΣ
Συνετή χρήση – εφαρμογή κανόνων χρήσης αντιβιοτικών Πότε; Σε ποιόν ασθενή; Ποιο ή ποια αντιβιοτικά Πώς; Για πόσο διάστημα; Έλεγχος –επιτήρηση λοιμώξεων (υγιεινή χεριών, έλεγχος, απομόνωση) 56
 56.")
59
The CDC has produced a number of clinical guidelines related the judicious use of antibiotics. To summarize these guidelines…

60
Κριτήρια Centor για στρεπτοκοκκική (GABHS ) φαρυγγοαμυγδαλίτιδα
ΚΛΑΣΙΚΑ ΚΡΙΤΗΡΙΑ ΤΡΟΠΟΠΟΙΗΜΕΝΗ ΒΑΘΜΟΛΟΓΙΑ Πυρετός (38οC) 1 βαθμός Τραχηλικοί λεμφαδένες 1 Εξίδρωμα αμυγδαλών 1 Απουσία βήχα (ρινίτιδας) 1 ΚΑΙ 1 για ηλικία 3-14 ετών 0 για ηλικία ετών -1 για ηλικία ετών Ι. Εάν θετικά 3 από 4 Πιθανότητα στρεπ. φαρυγγοαμ % ΙΙ. Εάν αρνητικά 3 από 4 Αρνητ. προγνωστική αξία GABHS 80% Βαθμολογία Αντιμετώπιση 1 Όχι έλεγχος Strep test > Θεραπεία ή Strep test 60
 1 βαθμός. Τραχηλικοί λεμφαδένες 1. Εξίδρωμα αμυγδαλών 1. Απουσία βήχα (ρινίτιδας) 1. ΚΑΙ. 1 για ηλικία 3-14 ετών. 0 για ηλικία ετών. -1 για ηλικία ετών. Ι. Εάν θετικά 3 από 4 Πιθανότητα στρεπ. φαρυγγοαμ % ΙΙ. Εάν αρνητικά 3 από 4 Αρνητ. προγνωστική αξία GABHS 80% Βαθμολογία Αντιμετώπιση. 1 Όχι έλεγχος. 2-3 Strep test. >4 Θεραπεία ή Strep test. 60.")
61
(17th Century, National Gallery, London)
61

63
ΚΑΤΑΛΛΗΛΗ ΧΡΗΣΗ ΑΝΤΙΒΙΟΤΙΚΩΝ
Έγκαιρη και επαρκής αντιμικροβιακή θεραπεία Αποφυγή περιττής χρήσης αντιβιοτικών This is the foundation of antimicrobial stewardship. Talk about studies that have increased mortality with inadequate initial therapy or delayed therapy. Then talk about how if you don’t de-escalate, youre putting your patient at a higher rate of having a drug-resistant infection in the future. In addition, decrease the chance of hospital-wide resistance and potential endemic situations. ↓ Θνητότητα ↓ Αντιμικροβιακή αντοχή 63

64
Η ΚΑΤΑΧΡΗΣΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΟ ΝΟΣΟΚΟΜΕΙΟ
Η ΚΑΤΑΧΡΗΣΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΟ ΝΟΣΟΚΟΜΕΙΟ Περίπου το 50% των χορηγούμενων αντιβιοτικών είναι ακατάλληλα ή περιττά Η ακατάλληλη χρήση αντιβιοτικών αποτελεί ιατρικό σφάλμα συμβάλλει στην ανάπτυξη ανθεκτικών βακτηρίων προκαλεί κολίτιδα από Clostridium difficile και άλλες ΑΕ Η μείωση κατανάλωσης αντίστασης και λοίμωξης από C. difficile Βασικοί κανόνες Καθορισμός ένδειξης, δοσολογίας και διάρκειας αντιμικροβιακής θεραπείας Λήψη καλλιέργειας Συμμόρφωση προς το αντιβιόγραμμα (αποκλιμάκωση, προσαρμογή, ή ?διακοπή)
")
65
Η ΚΑΤΑΧΡΗΣΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΗΝ ΚΤΗΝΟΤΡΟΦΙΑ
Η ΚΑΤΑΧΡΗΣΗ ΤΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΗΝ ΚΤΗΝΟΤΡΟΦΙΑ ΑΥΞΑΝΕΙ ΤΗΝ ΑΝΑΠΤΥΞΗ ΠΟΛΥΑΝΘΕΚΤΙΚΩΝ ΒΑΚΤΗΡΙΩΝ ΠΟΥ ΜΕΤΑΔΙΔΟΝΤΑΙ ΣΤΟΝ ΑΝΘΡΩΠΟ

66
Η ΑΝΑΚΑΛΥΨΗ ΝΕΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ ΠΕΡΙΟΡΙΖΕΤΑΙ Ή ΚΑΘΥΣΤΕΡΕΙ

69
ΑΝΤΙΜΙΚΡΟΒΙΑΚΗ ΘΕΡΑΠΕΙΑ
Αιτιολογική (ιδεώδης) Καλλιέργεια Αντιβιόγραμμα MIC (κυρίως σε μηνιγγίτιδα, ενδοκαρδίτιδα, μικροβιαιμία) Εμπειρική (βασίζεται σε κανόνες) Εν αναμονή αποτελεσμάτων Ευρέως φάσματος, προσαρμογή μετά το αντιβιόγραμμα Όταν δεν απομονώνεται το παθογόνο. 69
 Καλλιέργεια. Αντιβιόγραμμα. MIC (κυρίως σε μηνιγγίτιδα, ενδοκαρδίτιδα, μικροβιαιμία) Εμπειρική (βασίζεται σε κανόνες) Εν αναμονή αποτελεσμάτων. Ευρέως φάσματος, προσαρμογή μετά το αντιβιόγραμμα. Όταν δεν απομονώνεται το παθογόνο. 69.")
70
Minimal Inhibitory Concentration (MIC) vs.
Minimal Bactericidal Concentration (MBC) 32 ug/ml 16 ug/ml 8 ug/ml 4 ug/ml 2 ug/ml 1 ug/ml Sub-culture to agar medium MIC = 8 μg/ml MBC = 16 μg/ml
 32 ug/ml. 16 ug/ml. 8 ug/ml. 4 ug/ml. 2 ug/ml. 1 ug/ml. Sub-culture to agar medium. MIC = 8 μg/ml. MBC = 16 μg/ml.")
71
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών, προηγ. νοσηλείες) Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων (90% λοιμώξ. ενδογενείς) Φάσμα, γνώση γενικής και τοπικής αντοχής μικροβίων Αμυντικό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα – Κόστος Φαρμακοκινητική, φαρμακοδυναμική 71
 Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων (90% λοιμώξ. ενδογενείς) Φάσμα, γνώση γενικής και τοπικής αντοχής μικροβίων. Αμυντικό σύστημα ασθενούς. Ηλικία, Ιστορικό αλλεργίας. Γενετικές ή μεταβολικές διαταραχές. Κύηση ή θηλασμός. Νεφρική ή ηπατική ανεπάρκεια. Τοξικότητα – Κόστος. Φαρμακοκινητική, φαρμακοδυναμική. 71.")
72
ΒΑΚΤΗΡΙΑ ΑΝΑΛΟΓΑ ΜΕ ΘΕΣΗ ΛΟΙΜΩΞΗΣ
72

73
Προβληματικές περιοχές (κακή διείσδυση αντιβιοτικών)
Προστάτης Οφθαλμός Εκβλαστήσεις βαλβίδων Εγκεφαλονωτιαίο υγρό Αποστήματα Οστά Υγρό κυψελίδων πνεύμονα. 73

74
ΔΙΕΙΣΔΥΣΗ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΟ ΕΝΥ
Πολύ καλή Χλωραμφενικόλη Tριμεθοπρίμη - Σουλφαμεθοξαζόλη Μετρονιδαζόλη Νεότερες κινολόνες 74

75
ΔΙΕΙΣΔΥΣΗ ΑΝΤΙΒΙΟΤΙΚΩΝ ΣΤΟ ΕΝΥ
Καλή σε φλεγμονή των μηνίγγων Β λακταμικά Πενικιλλίνες, Κεφουροξίμη, Κεφαλοσπορίνες γ γενεάς, Μεροπενέμη, Αζτρεονάμη Βανκομυκίνη, Τεϊκοπλανίνη, Λινεζολίδη Ισονιαζίδη, Πυραζιναμίδη, Εθαμβουτόλη 5-Φθοριοκυτοσίνη, Αμφοτερικίνη Β. 75

76
Ενδοκυττάρια συγκέντρωση αντιβιοτικών
Ενδοκυττάρια παθογόνα Legionnella spp Mycoplasma (όχι πάντα) Chlamydiae Brucella Listeria monocytogene Salmonella Mycobacteria Meningococci Rhodococcus equi β-λακταμικά: <1 x Αμινογλυκοσίδες: <1-2 x Τετρακυκλίνες: 2-4 x Φθοριοκινολόνες: 5-20 x Μακρολίδες 4 έως 100 x
 Chlamydiae. Brucella. Listeria monocytogene. Salmonella. Mycobacteria. Meningococci. Rhodococcus equi. β-λακταμικά: <1 x. Αμινογλυκοσίδες: <1-2 x. Τετρακυκλίνες: 2-4 x. Φθοριοκινολόνες: 5-20 x. Μακρολίδες 4 έως 100 x.")
77
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών; προηγ. νοσηλείες;) Θέση της λοίμωξης Γνώση φυσιολογικής χλωρίδας (90% λοιμώξ. ενδογενείς) Φάσμα, γνώση γενικής και τοπικής αντοχής μικροβίων Αμυντικό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα – Κόστος Φαρμακοκινητική, φαρμακοδυναμική 77
 Θέση της λοίμωξης. Γνώση φυσιολογικής χλωρίδας (90% λοιμώξ. ενδογενείς) Φάσμα, γνώση γενικής και τοπικής αντοχής μικροβίων. Αμυντικό σύστημα ασθενούς. Ηλικία, Ιστορικό αλλεργίας. Γενετικές ή μεταβολικές διαταραχές. Κύηση ή θηλασμός. Νεφρική ή ηπατική ανεπάρκεια. Τοξικότητα – Κόστος. Φαρμακοκινητική, φαρμακοδυναμική. 77.")
78
Στείρες μικροβίων περιοχές
Μέσο ους, παραρρίνιοι κόλποι Λάρυγγας, τραχεία, βρόγχοι, βρογχιόλια, κυψελίδες Οισοφάγος, στόμαχος Κεντρικό τμήμα λεπτού εντέρου, ήπαρ, πάγκρεας, χοληφόρα Περιτοναϊκή κοιλότητα Μήτρα Νεφροί, Ουρητήρες, Ουροδόχος, Προστάτης, Ουρήθρα (εκτός προσθίας)
")
79
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών; νοσηλείες;) Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων (90% των λοιμώξεων ενδογενείς) Φάσμα, γνώση τοπικής αντοχής μικροβίων Ανοσιακό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Φαρμακοκινητική, φαρμακοδυναμική Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα - Κόστος. 79
 Θέση της λοίμωξης. Γνώση φυσιολογικών χλωρίδων (90% των λοιμώξεων ενδογενείς) Φάσμα, γνώση τοπικής αντοχής μικροβίων. Ανοσιακό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας. Φαρμακοκινητική, φαρμακοδυναμική. Γενετικές ή μεταβολικές διαταραχές. Κύηση ή θηλασμός. Νεφρική ή ηπατική ανεπάρκεια. Τοξικότητα - Κόστος. 79.")
80
ΑΝΟΣΟΛΟΓΙΚΕΣ ΔΙΑΤΑΡΑΧΕΣ ΚΑΙ ΛΟΙΜΩΞΕΙΣ
Έλλειψη C5-C9 Ναϊσέριες (N. meningitidis, N. gonorhoeae) Λειτουργική ή ανατομική έλλειψη σπλήνα Ελυτροφόρα βακτήρια πνευμονιόκοκκος, μηνιγγιτιδόκοκκος, αιμόφιλος γρίππης AIDS Μυκοβακτηρίδια, Pneumocystis jirovecii (πρώην carinii), Toxoplasma, ιοί, άλλοι μύκητες, άλλα πρωτόζωα. 80
 Λειτουργική ή ανατομική έλλειψη σπλήνα. Ελυτροφόρα βακτήρια. πνευμονιόκοκκος, μηνιγγιτιδόκοκκος, αιμόφιλος γρίππης. AIDS. Μυκοβακτηρίδια, Pneumocystis jirovecii (πρώην carinii), Toxoplasma, ιοί, άλλοι μύκητες, άλλα πρωτόζωα. 80.")
81
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών; νοσηλείες;) Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο Ανοσιακό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα - Κόστος. Φαρμακοκινητική, φαρμακοδυναμική 81
 Θέση της λοίμωξης. Γνώση φυσιολογικών χλωρίδων. Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο. Ανοσιακό σύστημα ασθενούς. Ηλικία, Ιστορικό αλλεργίας. Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός. Νεφρική ή ηπατική ανεπάρκεια. Τοξικότητα - Κόστος. Φαρμακοκινητική, φαρμακοδυναμική. 81.")
82
Αιμόλυση σε ένδεια G-6-PD
Σουλφοναμίδες Νιτροφουραντοΐνη Χλωραμφενικόλη Σουλφόνες Πριμακίνη Κινολόνες; 82

83
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών; νοσηλείες;) Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο Ανοσιακό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα - Κόστος. Φαρμακοκινητική, φαρμακοδυναμική 83
 Θέση της λοίμωξης. Γνώση φυσιολογικών χλωρίδων. Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο. Ανοσιακό σύστημα ασθενούς. Ηλικία, Ιστορικό αλλεργίας. Γενετικές ή μεταβολικές διαταραχές. Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια. Τοξικότητα - Κόστος. Φαρμακοκινητική, φαρμακοδυναμική. 83.")
84
ΑΝΤΙΒΙΟΤΙΚΑ ΚΑΤΑ ΤΗΝ ΚΥΗΣΗ
Ομάδα A: Μελέτες σε ανθρώπους δεν έδειξαν κίνδυνο για μητέρα ή έμβρυο Ομάδα Β: Μελέτες σε πειραματόζωα δεν έδειξαν τοξικότητα (ή έδειξαν, αλλά δεν έχει περιγραφεί τοξικότητα σε ανθρώπους) Ομάδα C: Μελέτες σε πειραματόζωα έδειξαν τοξικότητα, ανεπαρκή στοιχεία για άνθρωπο Ομάδα D: Στοιχεία για τοξικότητα σε άνθρωπο Ομάδα X: Ανακοινώθηκαν βλάβες στο έμβρυο σε ανθρώπους. ANTIBIOTICS IN PREGNANCY Section 9 of 10 Author Information Group B streptococcus< Em> Urinary Tract Infections Listeriosis Syphilis Chlamydia Gonorrhea Bacterial Vaginosis Antibiotics In Pregnancy Bibliography Cautious use of antibiotics is especially important during pregnancy because they can affect both the mother and the fetus. Without exception, antimicrobials cross the placenta; thus, the fetus is exposed to the adverse effects of every antibiotic taken by the mother. Although prescription drugs account for less than 1% of all congenital malformations, the preventability, if not the predictability, of the effects of antibiotic use make it a serious consideration. Classification Very few data are available regarding the teratogenic potential of most antibiotics in humans. The US Food and Drug Administration has categorized all antibiotics according to the risks associated with their use in pregnancy. These categories are as follows: Category A: Studies in pregnant women do not demonstrate any risks to the mother or fetus. Category B: While animal studies show no risk, human studies are inadequate or animal toxicity has been noted but the studies on humans show no risk. Category C: Animal studies indicate toxicity but human studies are inadequate. Category D: There is evidence of human risk. Category X: There have been reported fetal abnormalities in humans. In all classes of drugs, the benefits of antibiotic use must always outweigh the risks. Commonly used antibiotics Penicillins (category B): These are the most widely used antibiotics in pregnancy because of their wide margin of safety and lack of known toxicity. In the collaborative perinatal project, 3546 women used penicillin during the first trimester and no adverse effects were demonstrated. Ticarcillin, however, has shown some toxicity in animals and may not be safe in pregnancy. Cephalosporins (category B): This group has not been well studied in the first trimester and should therefore not be considered the first line of treatment in the first trimester of pregnancy. Generally, these drugs are considered safe and have shown no teratogenicity. Sulfonamides (category C): Avoid sulfonamides in the third trimester of pregnancy and during breastfeeding. Although these agents cause no known damage in utero, they can cause hyperbilirubinemia and kernicterus if the drug is still present in the neonate after birth. In mothers with G-6-PD deficiency, sulfonamide use has been associated with hemolysis. The combination of sulfonamides with trimethoprim in the first trimester has been associated with cardiovascular birth defects. Tetracyclines (category D): Tetracyclines have identifiable adverse effects in both the mother and the fetus. Pregnant women are susceptible to acute fatty necrosis of the liver, pancreatitis, and renal damage. In the fetus, these agents can cause stunting of growth, discoloration of teeth, and hypoplasia of dental enamel. Although tetracyclines have not proven to be deleterious in the first trimester or in smaller doses, they are best avoided. Aminoglycosides (category D): Aminoglycosides used in conjunction with hypomagnesemia and hypocalcemia, and in conjunction with calcium channel blockers, may cause neuromuscular blockade. Streptomycin is thus far the only agent in the class with reported toxicity, causing mild congenital deafness only detectable with vestibular testing or with an audiogram. Nitrofurantoin (category B): The collaborative perinatal project showed no increased risk of anomalies in 590 women who were exposed to the drug. In mothers with G-6-PD deficiency, it has caused hemolysis in both the mother and the fetus and should therefore be avoided near delivery. Quinolones (category C): Although animal studies have shown arthropathies, no human studies have been conducted and no cases of teratogenicity have been reported. These agents have a high affinity for bone and cartilage. Metronidazole (category B): Metronidazole should not be used in the first trimester or during lactation. When used in the second or third trimester, large single-dose treatments should be avoided. Macrolides (category B): These agents have not been associated with birth defects and are considered safe for use in pregnancy. Clindamycin (category B): This drug has not been associated with birth defects. 84
 Ομάδα C: Μελέτες σε πειραματόζωα έδειξαν τοξικότητα, ανεπαρκή στοιχεία για άνθρωπο. Ομάδα D: Στοιχεία για τοξικότητα σε άνθρωπο. Ομάδα X: Ανακοινώθηκαν βλάβες στο έμβρυο σε ανθρώπους. ANTIBIOTICS IN PREGNANCY. Section 9 of 10. Author Information Group B streptococcus< Em> Urinary Tract Infections Listeriosis Syphilis Chlamydia Gonorrhea Bacterial Vaginosis Antibiotics In Pregnancy. Bibliography. Cautious use of antibiotics is especially important during pregnancy because they can affect both the mother and the fetus. Without. exception, antimicrobials cross the placenta; thus, the fetus is exposed to the adverse effects of every antibiotic taken by the mother. Although prescription drugs account for less than 1% of all congenital malformations, the preventability, if not the predictability, of the. effects of antibiotic use make it a serious consideration. Classification. Very few data are available regarding the teratogenic potential of most antibiotics in humans. The US Food and Drug Administration. has categorized all antibiotics according to the risks associated with their use in pregnancy. These categories are as follows: Category A: Studies in pregnant women do not demonstrate any risks to the mother or fetus. Category B: While animal studies show no risk, human studies are inadequate or animal toxicity has been noted but the studies on. humans show no risk. Category C: Animal studies indicate toxicity but human studies are inadequate. Category D: There is evidence of human risk. Category X: There have been reported fetal abnormalities in humans. In all classes of drugs, the benefits of antibiotic use must always outweigh the risks. Commonly used antibiotics. Penicillins (category B): These are the most widely used antibiotics in pregnancy because of their wide margin of safety and lack. of known toxicity. In the collaborative perinatal project, 3546 women used penicillin during the first trimester and no adverse. effects were demonstrated. Ticarcillin, however, has shown some toxicity in animals and may not be safe in pregnancy. Cephalosporins (category B): This group has not been well studied in the first trimester and should therefore not be considered. the first line of treatment in the first trimester of pregnancy. Generally, these drugs are considered safe and have shown no. teratogenicity. Sulfonamides (category C): Avoid sulfonamides in the third trimester of pregnancy and during breastfeeding. Although these. agents cause no known damage in utero, they can cause hyperbilirubinemia and kernicterus if the drug is still present in the. neonate after birth. In mothers with G-6-PD deficiency, sulfonamide use has been associated with hemolysis. The combination of. sulfonamides with trimethoprim in the first trimester has been associated with cardiovascular birth defects. Tetracyclines (category D): Tetracyclines have identifiable adverse effects in both the mother and the fetus. Pregnant women are. susceptible to acute fatty necrosis of the liver, pancreatitis, and renal damage. In the fetus, these agents can cause stunting of. growth, discoloration of teeth, and hypoplasia of dental enamel. Although tetracyclines have not proven to be deleterious in the. first trimester or in smaller doses, they are best avoided. Aminoglycosides (category D): Aminoglycosides used in conjunction with hypomagnesemia and hypocalcemia, and in conjunction. with calcium channel blockers, may cause neuromuscular blockade. Streptomycin is thus far the only agent in the class with. reported toxicity, causing mild congenital deafness only detectable with vestibular testing or with an audiogram. Nitrofurantoin (category B): The collaborative perinatal project showed no increased risk of anomalies in 590 women who were. exposed to the drug. In mothers with G-6-PD deficiency, it has caused hemolysis in both the mother and the fetus and should. therefore be avoided near delivery. Quinolones (category C): Although animal studies have shown arthropathies, no human studies have been conducted and no. cases of teratogenicity have been reported. These agents have a high affinity for bone and cartilage. Metronidazole (category B): Metronidazole should not be used in the first trimester or during lactation. When used in the second. or third trimester, large single-dose treatments should be avoided. Macrolides (category B): These agents have not been associated with birth defects and are considered safe for use in pregnancy. Clindamycin (category B): This drug has not been associated with birth defects. 84.")
85
ΑΝΤΙΒΙΟΤΙΚΑ ΚΑΙ ΚΥΗΣΗ Ομάδα Β
Πενικιλλίνες (όχι τικαρσιλλίνη τοξική σε πειραματόζωα) Κεφαλοσπορίνες (ανεπαρκείς μελέτες για α τρίμηνο) Μακρολίδια Κλινδαμυκίνη Νιτροφοραντοϊνη (! έλλειψη G-6-PD) Μετρονιδαζόλη (όχι στο α τρίμηνο ή σε γαλουχία). ANTIBIOTICS IN PREGNANCY Section 9 of 10 Author Information Group B streptococcus< Em> Urinary Tract Infections Listeriosis Syphilis Chlamydia Gonorrhea Bacterial Vaginosis Antibiotics In Pregnancy Bibliography Cautious use of antibiotics is especially important during pregnancy because they can affect both the mother and the fetus. Without exception, antimicrobials cross the placenta; thus, the fetus is exposed to the adverse effects of every antibiotic taken by the mother. Although prescription drugs account for less than 1% of all congenital malformations, the preventability, if not the predictability, of the effects of antibiotic use make it a serious consideration. Classification Very few data are available regarding the teratogenic potential of most antibiotics in humans. The US Food and Drug Administration has categorized all antibiotics according to the risks associated with their use in pregnancy. These categories are as follows: Category A: Studies in pregnant women do not demonstrate any risks to the mother or fetus. Category B: While animal studies show no risk, human studies are inadequate or animal toxicity has been noted but the studies on humans show no risk. Category C: Animal studies indicate toxicity but human studies are inadequate. Category D: There is evidence of human risk. Category X: There have been reported fetal abnormalities in humans. In all classes of drugs, the benefits of antibiotic use must always outweigh the risks. Commonly used antibiotics Penicillins (category B): These are the most widely used antibiotics in pregnancy because of their wide margin of safety and lack of known toxicity. In the collaborative perinatal project, 3546 women used penicillin during the first trimester and no adverse effects were demonstrated. Ticarcillin, however, has shown some toxicity in animals and may not be safe in pregnancy. Cephalosporins (category B): This group has not been well studied in the first trimester and should therefore not be considered the first line of treatment in the first trimester of pregnancy. Generally, these drugs are considered safe and have shown no teratogenicity. Sulfonamides (category C): Avoid sulfonamides in the third trimester of pregnancy and during breastfeeding. Although these agents cause no known damage in utero, they can cause hyperbilirubinemia and kernicterus if the drug is still present in the neonate after birth. In mothers with G-6-PD deficiency, sulfonamide use has been associated with hemolysis. The combination of sulfonamides with trimethoprim in the first trimester has been associated with cardiovascular birth defects. Tetracyclines (category D): Tetracyclines have identifiable adverse effects in both the mother and the fetus. Pregnant women are susceptible to acute fatty necrosis of the liver, pancreatitis, and renal damage. In the fetus, these agents can cause stunting of growth, discoloration of teeth, and hypoplasia of dental enamel. Although tetracyclines have not proven to be deleterious in the first trimester or in smaller doses, they are best avoided. Aminoglycosides (category D): Aminoglycosides used in conjunction with hypomagnesemia and hypocalcemia, and in conjunction with calcium channel blockers, may cause neuromuscular blockade. Streptomycin is thus far the only agent in the class with reported toxicity, causing mild congenital deafness only detectable with vestibular testing or with an audiogram. Nitrofurantoin (category B): The collaborative perinatal project showed no increased risk of anomalies in 590 women who were exposed to the drug. In mothers with G-6-PD deficiency, it has caused hemolysis in both the mother and the fetus and should therefore be avoided near delivery. Quinolones (category C): Although animal studies have shown arthropathies, no human studies have been conducted and no cases of teratogenicity have been reported. These agents have a high affinity for bone and cartilage. Metronidazole (category B): Metronidazole should not be used in the first trimester or during lactation. When used in the second or third trimester, large single-dose treatments should be avoided. Macrolides (category B): These agents have not been associated with birth defects and are considered safe for use in pregnancy. Clindamycin (category B): This drug has not been associated with birth defects. 85
 Κεφαλοσπορίνες (ανεπαρκείς μελέτες για α τρίμηνο) Μακρολίδια. Κλινδαμυκίνη. Νιτροφοραντοϊνη (! έλλειψη G-6-PD) Μετρονιδαζόλη (όχι στο α τρίμηνο ή σε γαλουχία). ANTIBIOTICS IN PREGNANCY. Section 9 of 10. Author Information Group B streptococcus< Em> Urinary Tract Infections Listeriosis Syphilis Chlamydia Gonorrhea Bacterial Vaginosis Antibiotics In Pregnancy. Bibliography. Cautious use of antibiotics is especially important during pregnancy because they can affect both the mother and the fetus. Without. exception, antimicrobials cross the placenta; thus, the fetus is exposed to the adverse effects of every antibiotic taken by the mother. Although prescription drugs account for less than 1% of all congenital malformations, the preventability, if not the predictability, of the. effects of antibiotic use make it a serious consideration. Classification. Very few data are available regarding the teratogenic potential of most antibiotics in humans. The US Food and Drug Administration. has categorized all antibiotics according to the risks associated with their use in pregnancy. These categories are as follows: Category A: Studies in pregnant women do not demonstrate any risks to the mother or fetus. Category B: While animal studies show no risk, human studies are inadequate or animal toxicity has been noted but the studies on. humans show no risk. Category C: Animal studies indicate toxicity but human studies are inadequate. Category D: There is evidence of human risk. Category X: There have been reported fetal abnormalities in humans. In all classes of drugs, the benefits of antibiotic use must always outweigh the risks. Commonly used antibiotics. Penicillins (category B): These are the most widely used antibiotics in pregnancy because of their wide margin of safety and lack. of known toxicity. In the collaborative perinatal project, 3546 women used penicillin during the first trimester and no adverse. effects were demonstrated. Ticarcillin, however, has shown some toxicity in animals and may not be safe in pregnancy. Cephalosporins (category B): This group has not been well studied in the first trimester and should therefore not be considered. the first line of treatment in the first trimester of pregnancy. Generally, these drugs are considered safe and have shown no. teratogenicity. Sulfonamides (category C): Avoid sulfonamides in the third trimester of pregnancy and during breastfeeding. Although these. agents cause no known damage in utero, they can cause hyperbilirubinemia and kernicterus if the drug is still present in the. neonate after birth. In mothers with G-6-PD deficiency, sulfonamide use has been associated with hemolysis. The combination of. sulfonamides with trimethoprim in the first trimester has been associated with cardiovascular birth defects. Tetracyclines (category D): Tetracyclines have identifiable adverse effects in both the mother and the fetus. Pregnant women are. susceptible to acute fatty necrosis of the liver, pancreatitis, and renal damage. In the fetus, these agents can cause stunting of. growth, discoloration of teeth, and hypoplasia of dental enamel. Although tetracyclines have not proven to be deleterious in the. first trimester or in smaller doses, they are best avoided. Aminoglycosides (category D): Aminoglycosides used in conjunction with hypomagnesemia and hypocalcemia, and in conjunction. with calcium channel blockers, may cause neuromuscular blockade. Streptomycin is thus far the only agent in the class with. reported toxicity, causing mild congenital deafness only detectable with vestibular testing or with an audiogram. Nitrofurantoin (category B): The collaborative perinatal project showed no increased risk of anomalies in 590 women who were. exposed to the drug. In mothers with G-6-PD deficiency, it has caused hemolysis in both the mother and the fetus and should. therefore be avoided near delivery. Quinolones (category C): Although animal studies have shown arthropathies, no human studies have been conducted and no. cases of teratogenicity have been reported. These agents have a high affinity for bone and cartilage. Metronidazole (category B): Metronidazole should not be used in the first trimester or during lactation. When used in the second. or third trimester, large single-dose treatments should be avoided. Macrolides (category B): These agents have not been associated with birth defects and are considered safe for use in pregnancy. Clindamycin (category B): This drug has not been associated with birth defects. 85.")
86
ΑΝΤΙΒΙΟΤΙΚΑ ΚΑΙ ΚΥΗΣΗ Ομάδα C Ομάδα D Σουλφοναμίδες Κοτριμοξαζόλη
όχι σε γ τρίμηνο ή θηλασμό, πυρηνικός ίκτερος νεογνού Κοτριμοξαζόλη καρδιαγγειακές βλάβες εμβρύου σε α τρίμηνο Κινολόνες (αρθροπάθειες σε πειραματόζωα) Ομάδα D Τετρακυκλίνες οξεία ηπατική νέκρωση, παγκρεατίτιδα, νεφρική βλάβη αναστολή ανάπτυξης και βλάβες δοντιών εμβρύου Αμινογλυκοσίδες (στρεπτομυκίνη = ήπια κώφωση) ANTIBIOTICS IN PREGNANCY Cautious use of antibiotics is especially important during pregnancy because they can affect both the mother and the fetus. Without exception, antimicrobials cross the placenta; thus, the fetus is exposed to the adverse effects of every antibiotic taken by the mother. Although prescription drugs account for less than 1% of all congenital malformations, the preventability, if not the predictability, of the effects of antibiotic use make it a serious consideration. Classification Very few data are available regarding the teratogenic potential of most antibiotics in humans. The US Food and Drug Administration has categorized all antibiotics according to the risks associated with their use in pregnancy. These categories are as follows: Category A: Studies in pregnant women do not demonstrate any risks to the mother or fetus. Category B: While animal studies show no risk, human studies are inadequate or animal toxicity has been noted but the studies on humans show no risk. Category C: Animal studies indicate toxicity but human studies are inadequate. Category D: There is evidence of human risk. Category X: There have been reported fetal abnormalities in humans. In all classes of drugs, the benefits of antibiotic use must always outweigh the risks. Commonly used antibiotics Penicillins (category B): These are the most widely used antibiotics in pregnancy because of their wide margin of safety and lack of known toxicity. In the collaborative perinatal project, 3546 women used penicillin during the first trimester and no adverse effects were demonstrated. Ticarcillin, however, has shown some toxicity in animals and may not be safe in pregnancy. Cephalosporins (category B): This group has not been well studied in the first trimester and should therefore not be considered the first line of treatment in the first trimester of pregnancy. Generally, these drugs are considered safe and have shown no teratogenicity. Sulfonamides (category C): Avoid sulfonamides in the third trimester of pregnancy and during breastfeeding. Although these agents cause no known damage in utero, they can cause hyperbilirubinemia and kernicterus if the drug is still present in the neonate after birth. In mothers with G-6-PD deficiency, sulfonamide use has been associated with hemolysis. The combination of sulfonamides with trimethoprim in the first trimester has been associated with cardiovascular birth defects. Tetracyclines (category D): Tetracyclines have identifiable adverse effects in both the mother and the fetus. Pregnant women are susceptible to acute fatty necrosis of the liver, pancreatitis, and renal damage. In the fetus, these agents can cause stunting of growth, discoloration of teeth, and hypoplasia of dental enamel. Although tetracyclines have not proven to be deleterious in the first trimester or in smaller doses, they are best avoided. Aminoglycosides (category D): Aminoglycosides used in conjunction with hypomagnesemia and hypocalcemia, and in conjunction with calcium channel blockers, may cause neuromuscular blockade. Streptomycin is thus far the only agent in the class with reported toxicity, causing mild congenital deafness only detectable with vestibular testing or with an audiogram. Nitrofurantoin (category B): The collaborative perinatal project showed no increased risk of anomalies in 590 women who were exposed to the drug. In mothers with G-6-PD deficiency, it has caused hemolysis in both the mother and the fetus and should therefore be avoided near delivery. Quinolones (category C): Although animal studies have shown arthropathies, no human studies have been conducted and no cases of teratogenicity have been reported. These agents have a high affinity for bone and cartilage. Metronidazole (category B): Metronidazole should not be used in the first trimester or during lactation. When used in the second or third trimester, large single-dose treatments should be avoided. Macrolides (category B): These agents have not been associated with birth defects and are considered safe for use in pregnancy. Clindamycin (category B): This drug has not been associated with birth defects. 86
 Ομάδα D. Τετρακυκλίνες. οξεία ηπατική νέκρωση, παγκρεατίτιδα, νεφρική βλάβη. αναστολή ανάπτυξης και βλάβες δοντιών εμβρύου. Αμινογλυκοσίδες (στρεπτομυκίνη = ήπια κώφωση) ANTIBIOTICS IN PREGNANCY. Cautious use of antibiotics is especially important during pregnancy because they can affect both the mother and the fetus. Without. exception, antimicrobials cross the placenta; thus, the fetus is exposed to the adverse effects of every antibiotic taken by the mother. Although prescription drugs account for less than 1% of all congenital malformations, the preventability, if not the predictability, of the. effects of antibiotic use make it a serious consideration. Classification. Very few data are available regarding the teratogenic potential of most antibiotics in humans. The US Food and Drug Administration has categorized all antibiotics according to the risks associated with their use in pregnancy. These categories are as follows: Category A: Studies in pregnant women do not demonstrate any risks to the mother or fetus. Category B: While animal studies show no risk, human studies are inadequate or animal toxicity has been noted but the studies on humans show no risk. Category C: Animal studies indicate toxicity but human studies are inadequate. Category D: There is evidence of human risk. Category X: There have been reported fetal abnormalities in humans. In all classes of drugs, the benefits of antibiotic use must always outweigh the risks. Commonly used antibiotics. Penicillins (category B): These are the most widely used antibiotics in pregnancy because of their wide margin of safety and lack of known toxicity. In the collaborative perinatal project, 3546 women used penicillin during the first trimester and no adverse effects were demonstrated. Ticarcillin, however, has shown some toxicity in animals and may not be safe in pregnancy. Cephalosporins (category B): This group has not been well studied in the first trimester and should therefore not be considered the first line of treatment in the first trimester of pregnancy. Generally, these drugs are considered safe and have shown no teratogenicity. Sulfonamides (category C): Avoid sulfonamides in the third trimester of pregnancy and during breastfeeding. Although these agents cause no known damage in utero, they can cause hyperbilirubinemia and kernicterus if the drug is still present in the neonate after birth. In mothers with G-6-PD deficiency, sulfonamide use has been associated with hemolysis. The combination of sulfonamides with trimethoprim in the first trimester has been associated with cardiovascular birth defects. Tetracyclines (category D): Tetracyclines have identifiable adverse effects in both the mother and the fetus. Pregnant women are susceptible to acute fatty necrosis of the liver, pancreatitis, and renal damage. In the fetus, these agents can cause stunting of growth, discoloration of teeth, and hypoplasia of dental enamel. Although tetracyclines have not proven to be deleterious in the first trimester or in smaller doses, they are best avoided. Aminoglycosides (category D): Aminoglycosides used in conjunction with hypomagnesemia and hypocalcemia, and in conjunction with calcium channel blockers, may cause neuromuscular blockade. Streptomycin is thus far the only agent in the class with reported toxicity, causing mild congenital deafness only detectable with vestibular testing or with an audiogram. Nitrofurantoin (category B): The collaborative perinatal project showed no increased risk of anomalies in 590 women who were. exposed to the drug. In mothers with G-6-PD deficiency, it has caused hemolysis in both the mother and the fetus and should. therefore be avoided near delivery. Quinolones (category C): Although animal studies have shown arthropathies, no human studies have been conducted and no. cases of teratogenicity have been reported. These agents have a high affinity for bone and cartilage. Metronidazole (category B): Metronidazole should not be used in the first trimester or during lactation. When used in the second. or third trimester, large single-dose treatments should be avoided. Macrolides (category B): These agents have not been associated with birth defects and are considered safe for use in pregnancy. Clindamycin (category B): This drug has not been associated with birth defects. 86.")
87
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών; νοσηλείες;) Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο Ανοσιακό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα – Κόστος Φαρμακοκινητική, φαρμακοδυναμική 87
 Θέση της λοίμωξης. Γνώση φυσιολογικών χλωρίδων. Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο. Ανοσιακό σύστημα ασθενούς. Ηλικία, Ιστορικό αλλεργίας. Γενετικές ή μεταβολικές διαταραχές. Κύηση ή θηλασμός. Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα – Κόστος. Φαρμακοκινητική, φαρμακοδυναμική. 87.")
88
ΑΝΤΙΒΙΟΤΙΚΑ ΣΕ ΝΕΦΡΙΚΗ ΑΝΕΠΑΡΚΕΙΑ
Κανονική δοσολογία Ερυθρομυκίνη, κλινδαμυκίνη, χλωραμφενικόλη, δοξυκυκλίνη Αντισταφυλοκοκοκκικές πενικιλλίνες Κεφτριαξόνη Ριφαμπικίνη Ισονιαζίδη Μετρονιδαζόλη Λινεζολίδη 88

89
ΑΝΤΙΒΙΟΤΙΚΑ ΣΕ ΝΕΦΡΙΚΗ ΑΝΕΠΑΡΚΕΙΑ
Ελάττωση δόσης σε Ν.Α. Αμινογλυκοσίδες Βανκομυκίνη, τεϊκοπλανίνη Κοτριμοξαζόλη Κινολόνες Ιμιπενέμη Αζτρεονάμη Πενικιλλίνες Κεφαλοσπορίνες Εθαμβουτόλη. 89

90
ΑΝΤΙΒΙΟΤΙΚΑ ΣΕ ΝΕΦΡΙΚΗ ΑΝΕΠΑΡΚΕΙΑ
Αντενδείκνυνται Νιτροφουραντοΐνη Κεφαλοριδίνη Παρατεταμένης δράσεως σουλφοναμίδες Μεθεναμίνη ΠΑΣ. 90

91
ΠΡΟΣΟΧΗ ΣΕ ΗΠΑΤΙΚΗ ΑΝΕΠΕΑΡΚΕΙΑ
Ερυθρομυκίνη Μετρονιδαζόλη Κλινδαμυκίνη Χλωραμφενικόλη Τετρακυκλίνες Ριφαμπικίνη, Ισονιαζίδη, Πυραζιναμίδη Κετοκοναζόλη, φλουκοναζόλη Νιτροφουραντοΐνη Φουσιδικό οξύ. Chloramphenicol—higher risk of bone marrow suppression (markedly increased half life) Erythromycin estolate: causes cholestasis Tetracycline—dose related hepatotoxicity Antituberculous therapy in combinations, pyrazinamide Griseofulvin—contraindicated Nalidixic acid Nitrofurantoin prolonged use 91
 Erythromycin estolate: causes cholestasis. Tetracycline—dose related hepatotoxicity. Antituberculous therapy in combinations, pyrazinamide. Griseofulvin—contraindicated. Nalidixic acid. Nitrofurantoin prolonged use. 91.")
92
Antibiotics to be avoided in liver disease.
Chloramphenicol - higher risk of bone marrow suppression (markedly increased half life) Erythromycin estolate: causes cholestasis Tetracycline—dose related hepatotoxicity Antituberculous therapy in combinations, pyrazinamide Griseofulvin—contraindicated Nalidixic acid Nitrofurantoin prolonged use Deepak N. Amarapurkar. Prescribing Medications in Patients with Decompensated Liver Cirrhosis. International Journal of Hepatology (2011)
 Erythromycin estolate: causes cholestasis. Tetracycline—dose related hepatotoxicity. Antituberculous therapy in combinations, pyrazinamide. Griseofulvin—contraindicated. Nalidixic acid. Nitrofurantoin prolonged use. Deepak N. Amarapurkar. Prescribing Medications in Patients with Decompensated Liver Cirrhosis. International Journal of Hepatology (2011)")
93
Εμπειρική θεραπεία - Επιλογή αντιβιοτικού
Ιστορικό (+ λήψη αντιβιοτικών; νοσηλείες;) Θέση της λοίμωξης Γνώση φυσιολογικών χλωρίδων Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο Ανοσιακό σύστημα ασθενούς Ηλικία, Ιστορικό αλλεργίας Γενετικές ή μεταβολικές διαταραχές Κύηση ή θηλασμός Νεφρική ή ηπατική ανεπάρκεια Τοξικότητα – Κόστος Φαρμακοκινητική, φαρμακοδυναμική 93
 Θέση της λοίμωξης. Γνώση φυσιολογικών χλωρίδων. Φάσμα, γνώση αντοχής μικροβίων σε τοπικό επίπεδο. Ανοσιακό σύστημα ασθενούς. Ηλικία, Ιστορικό αλλεργίας. Γενετικές ή μεταβολικές διαταραχές. Κύηση ή θηλασμός. Νεφρική ή ηπατική ανεπάρκεια. Τοξικότητα – Κόστος. Φαρμακοκινητική, φαρμακοδυναμική. 93.")
94
ΦΑΡΜΑΚΟΚΙΝΗΤΙΚΗ ΑΝΤΙΒΙΟΤΙΚΩΝ Pharmacokinetic (PK)
Βιοδιαθεσιμότητα (f ή F) Επιφάνεια κάτω από καμπύλη Μέγιστη (Cmax), & ελάχιστη (Cmin συγκέντρωση ορού Όγκος κατανομής Σύνδεση με λευκώματα Ημιπερίοδος ζωής Μεταβολισμός ή βιομετατροπή Απέκκριση Κάθαρση (CL) PK parameters quantify the serum level time course of an antibiotic. The three pharmacokinetic parameters that are most important for evaluating antibiotic efficacy are the peak serum level (Cmax), the trough level (Cmin), and the Area Under the serum concentration time Curve (AUC). While these parameters quantify the serum level time course, they do not describe the killing activity of an antibiotic. Integrating the PK parameters with the MIC gives us three PK/PD parameters which quantify the activity of an antibiotic: the Peak/MIC ratio, the T>MIC, and the 24h-AUC/MIC ratio. The Peak/MIC ratio is simply the Cpmax divided by the MIC. The T>MIC (time above MIC) is the percentage of a dosage interval in which the serum level exceeds the MIC. The 24h-AUC/MIC ratio is determined by dividing the 24-hour-AUC by the MIC. Definition of the main pharmacokinetic parameters Pharmacokinetics is based on the study of the variation of plasma concentrations of drugs, because it is the only easily accessible parameter. Before approaching it, it is necessary to know the definitions of the terms usually used: The plasma half-life of a drug (T ½) is the time necessary to halve the plasma concentration, for example to decrease from 100 to 50 mg/L. The knowledge of the half-life is useful for the determination of the frequency of administration of a drug (the number of intakes per day) for obtaining the desired plasma concentration. Generally, the half-life of a particular drug is independent of the dose administered. In certain exceptional cases, it varies with the dose: it can increase or decrease according to, for example, the saturation of a mechanism (elimination, catabolism, binding to plasma proteins etc). The area under curve, AUC, corresponds to the integral of the plasma concentration versus an interval of definite time. In practice, the approximation is used: AUC = ƒ ([C] x Dt) [C]: is measured concentration and Dt: interval of time between two measurements. The precision of the AUC grows with the number of measurements of concentration taken. The AUC is expressed in mass (mg, g) X liter-1 X hour. One of its interests is to allow the measurement of the bioavailability of a drug. The bioavailability indicates the percentage of the administered drug which arrives in the central compartment. It is generally measured by comparing the AUC obtained after intravenous administration and after oral administration, for example. After intravenous administration, the AUC obtained corresponds to a bioavailability which, by definition, is 100%; after oral administration, the AUC corresponds at best to an identical bioavailability. It is generally lower, sometimes null. The compartment indicates the fictitious volume in which a drug would be distributed. It can correspond or not to a real volume, for example the volume of blood called first compartment, or the whole body except blood, called second compartment. The real anatomical sectors in which the drug is distributed at different concentrations is represented by one, two, rarely three virtual compartments where the concentration of the drug is regarded as homogeneous. The concept of compartment thus makes it possible to model the fate of a drug . The volume of distribution (Vd) is the fictitious volume, expressed in liter or in liter per kilogram, in which the drug would have been distributed by supposing that its concentration is homogeneous, i.e. the average tissue concentration is identical to that of the plasma. It is expressed as Vd = dose/C0 (initial concentration). For example, after intravenous injection of 100 mg of a drug whose initial concentration, C0, in plasma is 10 mg/L, the volume of distribution is of 10 L . For a given drug, the knowledge of its desirable concentration in blood and of its volume of distribution allows evaluation of the dose to administer. Clearance is the fraction of a theoretical volume completely purified (i.e. no longer containing any of the drug concerned) per unit of time. Plasma clearance is the apparent volume of plasma purified per unit of time. Total clearance (Clt) is the fraction of the volume of distribution, Vd, which is completely purified per unit of time. The total clearance depends on the constant of elimination and thus on T ½ and on Vd. Clearance is a constant in linear kinetics. Steady state concentration, Css, corresponds to the state of equilibrium obtained at the end of a certain number of administrations. To obtain an increase in the plasma concentration with repeated administrations, it is necessary that a residual concentration persists at the time of the following administration. At the steady state, if the dose and the frequency of administrations remain constant, the concentration obtained will also be constant. The steady state is obtained at the end of approximately five half-lives 94
 Επιφάνεια κάτω από καμπύλη. Μέγιστη (Cmax), & ελάχιστη (Cmin συγκέντρωση ορού. Όγκος κατανομής. Σύνδεση με λευκώματα. Ημιπερίοδος ζωής. Μεταβολισμός ή βιομετατροπή. Απέκκριση. Κάθαρση (CL) PK parameters quantify the serum level time course of an antibiotic. The three pharmacokinetic parameters that are most important for evaluating antibiotic efficacy are the peak serum level (Cmax), the trough level (Cmin), and the Area Under the serum concentration time Curve (AUC). While these parameters quantify the serum level time course, they do not describe the killing activity of an antibiotic. Integrating the PK parameters with the MIC gives us three PK/PD parameters which quantify the activity of an antibiotic: the Peak/MIC ratio, the T>MIC, and the 24h-AUC/MIC ratio. The Peak/MIC ratio is simply the Cpmax divided by the MIC. The T>MIC (time above MIC) is the percentage of a dosage interval in which the serum level exceeds the MIC. The 24h-AUC/MIC ratio is determined by dividing the 24-hour-AUC by the MIC. Definition of the main pharmacokinetic parameters. Pharmacokinetics is based on the study of the variation of plasma concentrations of drugs, because it is the only easily accessible parameter. Before approaching it, it is necessary to know the definitions of the terms usually used: The plasma half-life of a drug (T ½) is the time necessary to halve the plasma concentration, for example to decrease from 100 to 50 mg/L. The knowledge of the half-life is useful for the determination of the frequency of administration of a drug (the number of intakes per day) for obtaining the desired plasma concentration. Generally, the half-life of a particular drug is independent of the dose administered. In certain exceptional cases, it varies with the dose: it can increase or decrease according to, for example, the saturation of a mechanism (elimination, catabolism, binding to plasma proteins etc). The area under curve, AUC, corresponds to the integral of the plasma concentration versus an interval of definite time. In practice, the approximation is used: AUC = ƒ ([C] x Dt) [C]: is measured concentration and Dt: interval of time between two measurements. The precision of the AUC grows with the number of measurements of concentration taken. The AUC is expressed in mass (mg, g) X liter-1 X hour. One of its interests is to allow the measurement of the bioavailability of a drug. The bioavailability indicates the percentage of the administered drug which arrives in the central compartment. It is generally measured by comparing the AUC obtained after intravenous administration and after oral administration, for example. After intravenous administration, the AUC obtained corresponds to a bioavailability which, by definition, is 100%; after oral administration, the AUC corresponds at best to an identical bioavailability. It is generally lower, sometimes null. The compartment indicates the fictitious volume in which a drug would be distributed. It can correspond or not to a real volume, for example the volume of blood called first compartment, or the whole body except blood, called second compartment. The real anatomical sectors in which the drug is distributed at different concentrations is represented by one, two, rarely three virtual compartments where the concentration of the drug is regarded as homogeneous. The concept of compartment thus makes it possible to model the fate of a drug . The volume of distribution (Vd) is the fictitious volume, expressed in liter or in liter per kilogram, in which the drug would have been distributed by supposing that its concentration is homogeneous, i.e. the average tissue concentration is identical to that of the plasma. It is expressed as Vd = dose/C0 (initial concentration). For example, after intravenous injection of 100 mg of a drug whose initial concentration, C0, in plasma is 10 mg/L, the volume of distribution is of 10 L . For a given drug, the knowledge of its desirable concentration in blood and of its volume of distribution allows evaluation of the dose to administer. Clearance is the fraction of a theoretical volume completely purified (i.e. no longer containing any of the drug concerned) per unit of time. Plasma clearance is the apparent volume of plasma purified per unit of time. Total clearance (Clt) is the fraction of the volume of distribution, Vd, which is completely purified per unit of time. The total clearance depends on the constant of elimination and thus on T ½ and on Vd. Clearance is a constant in linear kinetics. Steady state concentration, Css, corresponds to the state of equilibrium obtained at the end of a certain number of administrations. To obtain an increase in the plasma concentration with repeated administrations, it is necessary that a residual concentration persists at the time of the following administration. At the steady state, if the dose and the frequency of administrations remain constant, the concentration obtained will also be constant. The steady state is obtained at the end of approximately five half-lives. 94.")
96
AUC= Area under curve, επιφάνεια κάτω από την καμπύλη
Definition of the main pharmacokinetic parameters Pharmacokinetics is based on the study of the variation of plasma concentrations of drugs, because it is the only easily accessible parameter. Before approaching it, it is necessary to know the definitions of the terms usually used: The plasma half-life of a drug (T ½) is the time necessary to halve the plasma concentration, for example to decrease from 100 to 50 mg/L. The knowledge of the half-life is useful for the determination of the frequency of administration of a drug (the number of intakes per day) for obtaining the desired plasma concentration. Generally, the half-life of a particular drug is independent of the dose administered. In certain exceptional cases, it varies with the dose: it can increase or decrease according to, for example, the saturation of a mechanism (elimination, catabolism, binding to plasma proteins etc). The area under curve, AUC, corresponds to the integral of the plasma concentration versus an interval of definite time. In practice, the approximation is used: AUC = ƒ ([C] x Dt) [C]: is measured concentration and Dt: interval of time between two measurements. The precision of the AUC grows with the number of measurements of concentration taken. The AUC is expressed in mass (mg, g) X liter-1 X hour. One of its interests is to allow the measurement of the bioavailability of a drug. The bioavailability indicates the percentage of the administered drug which arrives in the central compartment. It is generally measured by comparing the AUC obtained after intravenous administration and after oral administration, for example. After intravenous administration, the AUC obtained corresponds to a bioavailability which, by definition, is 100%; after oral administration, the AUC corresponds at best to an identical bioavailability. It is generally lower, sometimes null. The compartment indicates the fictitious volume in which a drug would be distributed. It can correspond or not to a real volume, for example the volume of blood called first compartment, or the whole body except blood, called second compartment. The real anatomical sectors in which the drug is distributed at different concentrations is represented by one, two, rarely three virtual compartments where the concentration of the drug is regarded as homogeneous. The concept of compartment thus makes it possible to model the fate of a drug . The volume of distribution (Vd) is the fictitious volume, expressed in liter or in liter per kilogram, in which the drug would have been distributed by supposing that its concentration is homogeneous, i.e. the average tissue concentration is identical to that of the plasma. It is expressed as Vd = dose/C0 (initial concentration). For example, after intravenous injection of 100 mg of a drug whose initial concentration, C0, in plasma is 10 mg/L, the volume of distribution is of 10 L . For a given drug, the knowledge of its desirable concentration in blood and of its volume of distribution allows evaluation of the dose to administer. Clearance is the fraction of a theoretical volume completely purified (i.e. no longer containing any of the drug concerned) per unit of time. Plasma clearance is the apparent volume of plasma purified per unit of time. Total clearance (Clt) is the fraction of the volume of distribution, Vd, which is completely purified per unit of time. The total clearance depends on the constant of elimination and thus on T ½ and on Vd. Clearance is a constant in linear kinetics. Steady state concentration, Css, corresponds to the state of equilibrium obtained at the end of a certain number of administrations. To obtain an increase in the plasma concentration with repeated administrations, it is necessary that a residual concentration persists at the time of the following administration. At the steady state, if the dose and the frequency of administrations remain constant, the concentration obtained will also be constant. The steady state is obtained at the end of approximately five half-lives AUC= Area under curve, επιφάνεια κάτω από την καμπύλη
 is the time necessary to halve the plasma concentration, for example to decrease from 100 to 50 mg/L. The knowledge of the half-life is useful for the determination of the frequency of administration of a drug (the number of intakes per day) for obtaining the desired plasma concentration. Generally, the half-life of a particular drug is independent of the dose administered. In certain exceptional cases, it varies with the dose: it can increase or decrease according to, for example, the saturation of a mechanism (elimination, catabolism, binding to plasma proteins etc). The area under curve, AUC, corresponds to the integral of the plasma concentration versus an interval of definite time. In practice, the approximation is used: AUC = ƒ ([C] x Dt) [C]: is measured concentration and Dt: interval of time between two measurements. The precision of the AUC grows with the number of measurements of concentration taken. The AUC is expressed in mass (mg, g) X liter-1 X hour. One of its interests is to allow the measurement of the bioavailability of a drug. The bioavailability indicates the percentage of the administered drug which arrives in the central compartment. It is generally measured by comparing the AUC obtained after intravenous administration and after oral administration, for example. After intravenous administration, the AUC obtained corresponds to a bioavailability which, by definition, is 100%; after oral administration, the AUC corresponds at best to an identical bioavailability. It is generally lower, sometimes null. The compartment indicates the fictitious volume in which a drug would be distributed. It can correspond or not to a real volume, for example the volume of blood called first compartment, or the whole body except blood, called second compartment. The real anatomical sectors in which the drug is distributed at different concentrations is represented by one, two, rarely three virtual compartments where the concentration of the drug is regarded as homogeneous. The concept of compartment thus makes it possible to model the fate of a drug . The volume of distribution (Vd) is the fictitious volume, expressed in liter or in liter per kilogram, in which the drug would have been distributed by supposing that its concentration is homogeneous, i.e. the average tissue concentration is identical to that of the plasma. It is expressed as Vd = dose/C0 (initial concentration). For example, after intravenous injection of 100 mg of a drug whose initial concentration, C0, in plasma is 10 mg/L, the volume of distribution is of 10 L . For a given drug, the knowledge of its desirable concentration in blood and of its volume of distribution allows evaluation of the dose to administer. Clearance is the fraction of a theoretical volume completely purified (i.e. no longer containing any of the drug concerned) per unit of time. Plasma clearance is the apparent volume of plasma purified per unit of time. Total clearance (Clt) is the fraction of the volume of distribution, Vd, which is completely purified per unit of time. The total clearance depends on the constant of elimination and thus on T ½ and on Vd. Clearance is a constant in linear kinetics. Steady state concentration, Css, corresponds to the state of equilibrium obtained at the end of a certain number of administrations. To obtain an increase in the plasma concentration with repeated administrations, it is necessary that a residual concentration persists at the time of the following administration. At the steady state, if the dose and the frequency of administrations remain constant, the concentration obtained will also be constant. The steady state is obtained at the end of approximately five half-lives. AUC= Area under curve, επιφάνεια κάτω από την καμπύλη.")
97
ΦΑΡΜΑΚΟΚΙΝΗΤΙΚΗ /ΦΑΡΜΑΚΟΔΥΝΑΜΙΚΗ ΑΝΤΙΒΙΟΤΙΚΩΝ
Peak (Peak/MIC) Συγκέντρωση στον ορό Area Under the Curve (AUC/MIC) Antimicrobial agents can be categorized on the basis of PK-PD parameters that is most predictive of efficacy. Based on experimental results, the 3 most common PK/PD parameters are: 1) Ratio of the maximum serum concentration (peak or Cmax) to the minimum inhibitory concentration (MIC); 2) ratio of the area under the plasma concentration versus time curve (AUC) versus MIC; and 3) the time during a dosing interval that plasma concentrations exceed the MIC. Another PD concept is the post-antibiotic effect which refers to persistent suppression of bacterial regrowth after brief exposure to an antibiotic. The duration of the PAE depends upon the particular antibiotic and pathogen involved. For example, there is little or no PAE against gram-negative bacteria with cell wall inhibitors such as beta-lactams. However, there is a greater PAE against gram-negative bacteria with antibiotics that inhibit protein or nucleic acid synthesis (AG, FQ). The PK-PD parameter that maps most closely with efficacy depends upon the bactericidal activity and the duration of the persistent effects. MIC Time above MIC PAE Time 97
 Συγκέντρωση στον ορό. Area Under the Curve (AUC/MIC) Antimicrobial agents can be categorized on the basis of PK-PD parameters that is most predictive of efficacy. Based on experimental results, the 3 most common PK/PD parameters are: 1) Ratio of the maximum serum concentration (peak or Cmax) to the minimum inhibitory concentration (MIC); 2) ratio of the area under the plasma concentration versus time curve (AUC) versus MIC; and 3) the time during a dosing interval that plasma concentrations exceed the MIC. Another PD concept is the post-antibiotic effect which refers to persistent suppression of bacterial regrowth after brief exposure to an antibiotic. The duration of the PAE depends upon the particular antibiotic and pathogen involved. For example, there is little or no PAE against gram-negative bacteria with cell wall inhibitors such as beta-lactams. However, there is a greater PAE against gram-negative bacteria with antibiotics that inhibit protein or nucleic acid synthesis (AG, FQ). The PK-PD parameter that maps most closely with efficacy depends upon the bactericidal activity and the duration of the persistent effects. MIC. Time above MIC. PAE. Time. 97.")
98
Φαρμακοδυναμική αντιβιοτικών (PD)
Υποδηλώνει τη σχέση φαρμακοκινητικών παραμέτρων με αντιμικροβιακή δράση αντιβιοτικού Cmax/MIC T>MIC, 24h-AUC/MIC

99
Φαρμακοδυναμική αντιβιοτικών (PD)
Cmax Cmax : MIC T > MIC AUC/MIC AUC Συγκέντρωση μg/ml MIC T>MIC Χρόνος Υποδηλώνει σχέση φαρμακοκινητικών παραμέτρων με αντιμικροβιακή δράση

100
TIME DEPENDENT ANTIBIOTICS
Χρονοεξαρτώμενη, T > MIC AUC MIC T > MIC Drug concentration Time Time-dependent antibiotics kill bacteria at the same rate and to the same extent once an appropriate antibiotic threshold concentration has been achieved. Increasing the antibiotic concentration beyond this point typically does not augment the antibacterial activity. For Time dependent antibiotics, how long the antibiotic will remain above MIC is important.

101
TIME DEPENDENT KILLING
Dosing interval : 12 hours Drug B Drug A DRUG B is More EFFICACIOUS : T > MIC = 90 % of Dosing Interval Concentration For example ;As Compare to Drug A, Drug B remain above the MIC for atleast ,10 hours thus , DRUG B is More EFFICACIOUS MIC 2 4 6 8 10 12 Time

102
PK/PD και αντιμικροβιακή δράση
Δραστικότητα των αντιβιοτικών: 1. Δοσοεξεαρτώμενη (Concentration dependent) Cmax/ MIC=10-12 Aminoglycosides, quinolones, daptomycine 2. Χρονοεξαρτώμεη (Time dependent) Συγκέντρωση > MIC στο 40-50% του μεσοδιαστήματος χορήγησης β – λακταμικά, γλυκοπεπτίδια, μακρολίδες 102
 Cmax/ MIC= Aminoglycosides, quinolones, daptomycine. 2. Χρονοεξαρτώμεη (Time dependent) Συγκέντρωση > MIC στο 40-50% του μεσοδιαστήματος χορήγησης. β – λακταμικά, γλυκοπεπτίδια, μακρολίδες")
103
CONCENTRATION DEPENDENT ANTIBIOTICS Δοσοεξαρτώμενα αντιβιοτικά: Cmax/MIC
A αποτελεσματικότερο Β, διότι επιτυγχάνει υψηλότερες συγκεντρώσεις > MIC Concentration Αντιβ. B For Example, Suppose there are two Drugs viz ;Drug A and Drug B. Compare to Drug B, Drug A Achieves higher concentration above the MIC ,thus Drug A is more Efficacious as compared to Drug B. MIC 2 4 6 8 10 12 Time

104
Χρονοεξαρτώμενη (Time dependent) δραστικότητα
A αποτελεσματικότερο Β, διότι επιτυγχάνει συγκεντρώσεις άνωθεν του MIC για το 50% του μεσοδιαστήματος χορήγησης 104

105
ΣΩΣΤΟΣ ΤΡΟΠΟΣ ΧΟΡΗΓΗΣΗΣ ΧΡΟΝΟΕΞΕΡΤΩΜΕΝΟΥ ΑΝΤΙΒΙΟΤΙΚΟΥ

106
Pharmacodynamics of a time-dependent antimicrobial. Shown is a
comparison of the simulated drug concentration profile of a timedependent anitmicrobial with an elimination half-life of 1 hour administered over 30 minutes or over 3 hours. The extended infusion time increases the time for which the antibiotic concentration exceeds the minimum inhibitory concentration (t>MIC). Dotted line refers to a MIC of 8 mg/l.
. Dotted line refers to a. MIC of 8 mg/l.")
107
Pharmacodynamics of a concentration-dependent antimicrobial.
Shown is a comparison of the simulated drug concentration profile of a concentration-dependent antimicrobial with an elimination half-life of 2 hours administered once daily or in two divided doses. Under the same total daily dose, once daily administration ensures higher Cmax/MIC ratio in presence of equal AUC/MIC ratio. Dotted line refers to a MIC of 2 mg/l. AUC, area under the plasma concentration-time curve; Cmax, peak plasma concentration; MIC, minimum inhibitory concentration

108
MEROPENEM 1 g bolus 2 g σε 3ωρη έγχυση 1 g σε 3ωρη έγχυση
Jaruratanasirikul et al AAC 2005;49(4):1337 108
:")
109
Τρόπος δραστικότητας αντιβιοτικού Τρόπος χορήγησης αντιβιοτικού
ΘΕΡΑΠΕΙΑ ΜΕ ΒΑΣΗ PD Τρόπος δραστικότητας αντιβιοτικού Αντιβιοτικά Τρόπος χορήγησης αντιβιοτικού Δοσοεξερτώμενη δράση Concentration dependent Αμινογλυκοσίδες Κινολόνες Δαπτομυκίνη Ημερήσια δόση εφάπαξ AUC/MIC, Cmax /MIC Χρονοεξαρτώμενη δράση Time dependent Β λακταμικά Κλινδαμυκίνη Μακρολίδες Μικρά μεσοδιαστήματα ή συνεχής έγχυση Τmax (T>MIC) Μικτή (κυρίως time dependent και λιγότερο concentration dependent) Γλυκοπεπτίδια, αζιθρο-μυκίνη, τετρακυκλίνες, στρεπτογραμμίνες, λινεζολίδη AUC/MIC 109
 Μικτή (κυρίως time dependent και λιγότερο concentration dependent) Γλυκοπεπτίδια, αζιθρο-μυκίνη, τετρακυκλίνες, στρεπτογραμμίνες, λινεζολίδη. AUC/MIC")
110
Διάρκεια αντιμικροβιακής θεραπείας
Στρεπτ. φαρυγγίτιδα: πενικιλλίνη, 10 ημ. Λοιμώξεις ουροποιητικού κατώτερου σε γυναίκες: 3-5 ημ. ή εφάπαξ. ανώτερου: (8) ημ. Πνευμονία κοινότητας και νοσοκομειακή: 8 ημ. Μικροβιακή μηνιγγίτιδα: H. influenzae, N. meningitidis: 7 ημ. S. pneumoniae: ημ. L. monocytogenes, εντεροβακτηριακά: >3 εβδ. Οξεία οστεομυελίτιδα ή ενδοκαρδίτιδα: 4-6 εβδ. Άλλες οξείες λοιμώξεις: 5-7 ημ. μετά την απυρεξία; 110
 ημ. Πνευμονία κοινότητας και νοσοκομειακή: 8 ημ. Μικροβιακή μηνιγγίτιδα: H. influenzae, N. meningitidis: 7 ημ. S. pneumoniae: ημ. L. monocytogenes, εντεροβακτηριακά: >3 εβδ. Οξεία οστεομυελίτιδα ή ενδοκαρδίτιδα: 4-6 εβδ. Άλλες οξείες λοιμώξεις: 5-7 ημ. μετά την απυρεξία; 110.")
111
ΚΛΙΝΙΚΗ ΚΑΤΑΤΑΞΗ ΑΝΤΙΒΙΟΤΙΚΩΝ
Πενικιλλίνες Κεφαλοσπορίνες Καρβαπενέμες Μονοβακτάμες Αμινογλυκοσίδες Κινολόνες Βανκομυκίνη, Τεϊκοπλανίνη Οξαζολιδίνες Λιποπεπτίδια Στρεπτογραμμίνες Πολυμυξίνες (A-E) Κετολίδες Μακρολίδια Λινκοσαμίνες Tετρακυκλίνες, Τιγκεκυκλίνη Χλωραμφενικόλη Ριφαμυκίνες Αναστ. σύνθεσης φυλλικού οξέος Φουσιδικό οξύ Νιτροϊμιδαζόλες Νιτροφουραντοϊνη Ισονιαζίδη, πυραζιναμίδη αιθαμβουτόλη, κυκλοσερίνη κ.ά.
 Κετολίδες. Μακρολίδια. Λινκοσαμίνες. Tετρακυκλίνες, Τιγκεκυκλίνη. Χλωραμφενικόλη. Ριφαμυκίνες. Αναστ. σύνθεσης φυλλικού οξέος. Φουσιδικό οξύ. Νιτροϊμιδαζόλες. Νιτροφουραντοϊνη. Ισονιαζίδη, πυραζιναμίδη αιθαμβουτόλη, κυκλοσερίνη κ.ά.")
112
β-ΛΑΚΤΑΜΙΚΑ ΑΝΤΙΒΙΟΤΙΚΑ
Πενικιλλίνες Κεφαλοσπορίνες Μονοβακτάμες Καρβαπενέμες

114
ΜΗΧΑΝΙΣΜΟΣ ΔΡΑΣΗΣ β-ΛΑΚΤΑΜΙΚΩΝ ΑΝΤΙΒΙΟΤΙΚΩΝ
Σύνδεση Ο=C-N με PPB (penicillin-binding proteins) PBP1a, PBP1b, PBP2, PBP3, PBP4 Αναστολή σύνθεσης πεπτιδογλυκάνης καταστροφή κυτ. τοιχώματος
 PBP1a, PBP1b, PBP2, PBP3, PBP4. Αναστολή σύνθεσης πεπτιδογλυκάνης καταστροφή κυτ. τοιχώματος.")
115
ΜΗΧΑΝΙΣΜΟΣ ΔΡΑΣΗΣ β-ΛΑΚΤΑΜΙΚΩΝ
Η πεπτιδογλυκάνη αποτελείται κυρίως από τα αμινοσάκχαρα Ν-ακετυλομουραμικό οξύ (ΝΑΜ) Ν-ακετυλογλυκοζαμίνη (NAG) Γειτονικά μόρια ΝΑΜ συνδέονται μεταξύ τους με πεπτιδικές αλυσίδες με τη βοήθεια τρανσεπεπτιδασών (cell wall transamidase –CWT- ή penicillin-binding proteins –PBP-).
 Ν-ακετυλογλυκοζαμίνη (NAG) Γειτονικά μόρια ΝΑΜ συνδέονται μεταξύ τους με πεπτιδικές αλυσίδες με τη βοήθεια τρανσεπεπτιδασών (cell wall transamidase –CWT- ή penicillin-binding proteins –PBP-).")
116
Τα β λακταμικά αντιβιοτικά μοιάζουν με τις πεπτιδικές αλυσίδες και συνδέονται με PBP αναστέλλουν τη σύνθεση της πεπτιδογλυκάνης εξασθενεί το κυτταρικό τοίχωμα λύση

117
Η ΕΠΙΔΡΑΣΗ ΤΩΝ Β-ΛΑΚΤΑΜΙΚΩΝ ΣΤΑ ΒΑΚΤΗΡΙΑ
S. aureus S. aureus μετά έκθεση σε penicillin G Giesbrecht et al., 1998, Microbiology and Molecular Biology Reviews 62:

118
MΗXΑΝΙΣΜΟΙ ΑΝΤΙΣΤΑΣΗΣ ΣΤΑ β-ΛΑΚΤΑΜΙΚΑ
Παραγωγή β-λακταμάσης (5b) Ελάττωση διαπερατότητας (πορίνες) κυτ. τοιχώματος (3) ή ενεργοποίηση αντλιών εκροής (π.χ. ψευδομονάδα) (6) Αλλαγή PPB (π.χ. MRSA, πνευμονιόκοκκος, γονόκοκκος κ.ά) (5a). Bacteria develop resistance to β-lactam antibiotics by a variety of mechanisms. Most common is the destruction of the drug by -lactamases. The -lactamases of gram-negative bacteria are confined to the periplasm, between the inner and outer membranes, while gram-positive bacteria secrete their -lactamases into the surrounding medium. These enzymes have a higher affinity for the antibiotic than the antibiotic has for its target. Binding results in hydrolysis of the -lactam ring. Genes encoding -lactamases have been found in both chromosomal and extrachromosomal locations and in both gram-positive and gram-negative bacteria; these genes are often on mobile genetic elements. Many "advanced-generation" -lactam antibiotics, such as ceftriaxone and cefepime, are stable in the presence of plasmid-mediated -lactamases and are active against bacteria resistant to earlier-generation -lactam antibiotics. However, extended-spectrum -lactamases (ESBLs), either acquired on mobile genetic elements by gram-negative bacteria (e.g., Klebsiella pneumoniae and Escherichia coli) or present as stable chromosomal genes in other gram-negative species (e.g., Enterobacter spp.), have broad substrate specificity, hydrolyzing virtually all penicillins and cephalosporins. One strategy that has been devised for circumventing resistance mediated by -lactamases is to combine the -lactam agent with an inhibitor that avidly binds the inactivating enzyme, preventing its attack on the antibiotic. Unfortunately, the inhibitors (e.g., clavulanic acid, sulbactam, and tazobactam) do not bind all chromosomal -lactamases (e.g., that of Enterobacter) and thus cannot be depended on to prevent the inactivation of -lactam antibiotics by such enzymes. No -lactam antibiotic or inhibitor has been produced that can resist all of the many -lactamases that have been identified. A second mechanism of bacterial resistance to -lactam antibiotics is an alteration in PBP targets so that the PBPs have a markedly reduced affinity for the drug. While this alteration may occur by mutation of existing genes, the acquisition of new PBP genes (as in staphylococcal resistance to methicillin) or of new pieces of PBP genes (as in streptococcal, gonococcal, and meningococcal resistance to penicillin) is more important. A final resistance mechanism is the coupling, in gram-negative bacteria, of a decrease in outer-membrane permeability with rapid efflux of the antibiotic from the periplasm to the cell exterior. Mutations of genes encoding outer-membrane protein channels called porins decrease the entry of -lactam antibiotics into the cell, while additional proteins form channels that actively pump -lactams out of the cell. Resistance of Enterobacteriaceae to some cephalosporins and resistance of Pseudomonas spp. to cephalosporins and piperacillin are the best examples of this mechanism.
 Ελάττωση διαπερατότητας (πορίνες) κυτ. τοιχώματος (3) ή ενεργοποίηση αντλιών εκροής (π.χ. ψευδομονάδα) (6) Αλλαγή PPB (π.χ. MRSA, πνευμονιόκοκκος, γονόκοκκος κ.ά) (5a). Bacteria develop resistance to β-lactam antibiotics by a variety of mechanisms. Most common is the destruction of the drug by -lactamases. The -lactamases of gram-negative bacteria are confined to the periplasm, between the inner and outer membranes, while gram-positive bacteria secrete their -lactamases into the surrounding medium. These enzymes have a higher affinity for the antibiotic than the antibiotic has for its target. Binding results in hydrolysis of the -lactam ring. Genes encoding -lactamases have been found in both chromosomal and extrachromosomal locations and in both gram-positive and gram-negative bacteria; these genes are often on mobile genetic elements. Many advanced-generation -lactam antibiotics, such as ceftriaxone and cefepime, are stable in the presence of plasmid-mediated -lactamases and are active against bacteria resistant to earlier-generation -lactam antibiotics. However, extended-spectrum -lactamases (ESBLs), either acquired on mobile genetic elements by gram-negative bacteria (e.g., Klebsiella pneumoniae and Escherichia coli) or present as stable chromosomal genes in other gram-negative species (e.g., Enterobacter spp.), have broad substrate specificity, hydrolyzing virtually all penicillins and cephalosporins. One strategy that has been devised for circumventing resistance mediated by -lactamases is to combine the -lactam agent with an inhibitor that avidly binds the inactivating enzyme, preventing its attack on the antibiotic. Unfortunately, the inhibitors (e.g., clavulanic acid, sulbactam, and tazobactam) do not bind all chromosomal -lactamases (e.g., that of Enterobacter) and thus cannot be depended on to prevent the inactivation of -lactam antibiotics by such enzymes. No -lactam antibiotic or inhibitor has been produced that can resist all of the many -lactamases that have been identified. A second mechanism of bacterial resistance to -lactam antibiotics is an alteration in PBP targets so that the PBPs have a markedly reduced affinity for the drug. While this alteration may occur by mutation of existing genes, the acquisition of new PBP genes (as in staphylococcal resistance to methicillin) or of new pieces of PBP genes (as in streptococcal, gonococcal, and meningococcal resistance to penicillin) is more important. A final resistance mechanism is the coupling, in gram-negative bacteria, of a decrease in outer-membrane permeability with rapid efflux of the antibiotic from the periplasm to the cell exterior. Mutations of genes encoding outer-membrane protein channels called porins decrease the entry of -lactam antibiotics into the cell, while additional proteins form channels that actively pump -lactams out of the cell. Resistance of Enterobacteriaceae to some cephalosporins and resistance of Pseudomonas spp. to cephalosporins and piperacillin are the best examples of this mechanism.")
119
ANEΠΙΘΥΜΗΤΕΣ ΕΝΕΡΓΕΙΕΣ β ΛΑΚΤΑΜΙΚΩΝ
Αλλεργικές αντιδράσεις. Γαστρεντερικές διαταραχές Διαταραχές αιμόστασης Ουδετεροπενία, αιμολυτική αναιμία, θρομβοπενία Νευροτοξικότητα Ηπατοτοξικότητα Νεφροτοξικότητα. Υποκαλιαιμική αλκάλωση Υπερκαλιαιμία ή υπερνατριαιμία Frequent: allergic reactions, rarely anaphylaxis, erythema multiforme or Stevens Johnson syndrome; rash (more common with ampicillin and amoxicillin than with other penicillins); diarrhea (most common with ampicillin and amoxicillin/clavulanic acid); nausea and vomiting with amoxicillin/clavulanic acid Occasional: hemolytic anemia; neutropenia; pseudomembranous colitis; platelet dysfunction with high doses of piperacillin, ticarcillin, nafcillin, or methicillin; cholestatic hepatitis with amoxicillin/ clavulanic acid Rare: hepatic damage with semisynthetic penicillins; granulocytopenia or agranulocytosis with semisynthetic penicillins; damage with semisynthetic penicillins and penicillin G; muscle irritability and seizures, usually after high doses in patients with impaired renal function; hyperkalemia and arrhythmias with IV potassium penicillin G given rapidly; bleeding diathesis; Henoch-Schönlein purpura with ampicillin; thrombocytopenia with methicillin and mezlocillin; terror, hallucinations, disorientation, agitation, bizarre behavior and neurological reactions with high doses of procaine penicillin G, oxacillin, or ticarcillin; hypokalemic alkalosis and/or sodium overload with high doses of ticarcillin or nafcillin; hemorrhagic cystitis with methicillin; GI bleeding with dicloxacillin; tissue damage with extravasation of nafcillin 119
; diarrhea (most common with ampicillin. and amoxicillin/clavulanic acid); nausea. and vomiting with amoxicillin/clavulanic. acid. Occasional: hemolytic anemia; neutropenia; pseudomembranous colitis; platelet. dysfunction with high doses of piperacillin, ticarcillin, nafcillin, or methicillin; cholestatic hepatitis with amoxicillin/ clavulanic acid. Rare: hepatic damage with semisynthetic. penicillins; granulocytopenia or agranulocytosis. with semisynthetic penicillins; damage with semisynthetic penicillins. and penicillin G; muscle irritability. and seizures, usually after high doses in. patients with impaired renal function; hyperkalemia and arrhythmias with IV. potassium penicillin G given rapidly; bleeding diathesis; Henoch-Schönlein purpura. with ampicillin; thrombocytopenia. with methicillin and mezlocillin; terror, hallucinations, disorientation, agitation, bizarre behavior and neurological. reactions with high doses of procaine. penicillin G, oxacillin, or ticarcillin; hypokalemic alkalosis and/or sodium. overload with high doses of ticarcillin or. nafcillin; hemorrhagic cystitis with methicillin; GI bleeding with dicloxacillin; tissue. damage with extravasation of nafcillin")
120
Αλλεργικές αντιδράσεις στα β-λακταμικά αντιβιοτικά (1-10%)
Οξεία αναφυλακτική αντίδραση Επιταχυνόμενη αλλεργική αντίδραση Δερματικό εξάνθημα Ορονοσία Αγγειίτιδα Πυρετός Εωσινοφιλία Οξεία διάμεση νεφρίτιδα Θρομβοπενική πορφύρα (σπάνια)
")
121
Οξεία αναφυλακτική αντίδραση
Συχνότητα: 0,02% (2 / ασθενείς που έλαβαν πενικιλίνη) Άμεση αντίδραση (2-60 min) * Ερύθημα, κνησμός, κνίδωση * Ρινίτιδα, βρογχόσπασμος * Υπόταση, Shock Θεραπεία: αδρεναλίνη * 0,3-0,5mg s.c. ή i.m., επανάληψη σε min * 0,3-0,5mg i.v., επανάληψη σε 5-10 min Θνητότητα: 10% 10-20 θάνατοι / ασθενείς που έλαβαν πενικιλλίνη
 Άμεση αντίδραση (2-60 min) * Ερύθημα, κνησμός, κνίδωση. * Ρινίτιδα, βρογχόσπασμος. * Υπόταση, Shock. Θεραπεία: αδρεναλίνη. * 0,3-0,5mg s.c. ή i.m., επανάληψη σε min. * 0,3-0,5mg i.v., επανάληψη σε 5-10 min. Θνητότητα: 10% θάνατοι / ασθενείς που έλαβαν πενικιλλίνη.")
122
Επιταχυνόμενη αλλεργική αντίδραση
Σε 1-72 ώρες μετά από χορήγηση β λακταμικού: Ερύθημα, Κνησμός Κνίδωση Ρινίτιδα, βρογχόσπασμος Οίδημα λάρυγγα.

123
Διάγνωση αλλεργίας στην πενικιλίνη
Ιστορικό Δοκιμασία σκαριφισμών (Scratch test) *Διάλυμα πολυλυσίνης + πενικιλλοϋλο-ομάδες (Prepen®) * Παρακολούθηση λεπτά * Διήθηση < 4mm ; Ενδοδερμική δοκιμασία ΕΔ ένεση 0.02 ml διαλύματος Διήθηση >4mm & ερυθρότητα >10 mm θετική δοκιμασία PRE-PEN® Penicillin Skin Test Antigen PRE-PEN is the only FDA approved skin test for the diagnosis of penicillin allergy. PRE-PEN is administered through both scratch and intradermal testing and quickly identifies patients who can safely receive penicillin. It is estimated that penicillin allergy is overstated due to non-confirmed, self-reported patient history. Approximately 85% of patients who describe themselves as penicillin allergic will have negative skin tests and can safely receive penicillin and related antibiotics. Confirmation of the presence of a true allergy is critical and non-confirmed penicillin allergy may result in: Unnecessary denial of an effective and well tolerated treatment Increased out of pocket costs for patients, since many alternatives to penicillins are Tier 3 medications Overuse of broad spectrum antibiotics leading to increased drug-resistant bacteria Visit to learn more
 *Διάλυμα πολυλυσίνης + πενικιλλοϋλο-ομάδες (Prepen®) * Παρακολούθηση λεπτά. * Διήθηση < 4mm ; Ενδοδερμική δοκιμασία. ΕΔ ένεση 0.02 ml διαλύματος. Διήθηση >4mm & ερυθρότητα >10 mm θετική δοκιμασία. PRE-PEN® Penicillin Skin Test Antigen. PRE-PEN is the only FDA approved skin test for the diagnosis of penicillin allergy. PRE-PEN is administered through both scratch and intradermal testing and quickly identifies patients who can safely receive penicillin. It is estimated that penicillin allergy is overstated due to non-confirmed, self-reported patient history. Approximately 85% of patients who describe themselves as penicillin allergic will have negative skin tests and can safely receive penicillin and related antibiotics. Confirmation of the presence of a true allergy is critical and non-confirmed penicillin allergy may result in: Unnecessary denial of an effective and well tolerated treatment. Increased out of pocket costs for patients, since many alternatives to penicillins are Tier 3 medications. Overuse of broad spectrum antibiotics leading to increased drug-resistant bacteria. Visit to learn more.")
124
ΠΕΝΙΚΙΛΙΝΕΣ Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V
* Πενικιλίνες περιορισμένου φάσματος Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V * Ανθεκτικές στην πενικιλινάση πενικιλίνες Μεθικιλλίνη, οξα-, δικλο-, κλοξα- φλουκλοξα, ναφ-κιλλίνη * Πενικιλίνες ευρέος φάσματος Αμινοπενικιλίνες (αμπικιλλίνη, αμοξυκιλίνη) Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη Penicillin G Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections
 Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης. αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ. τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη. Penicillin G. Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections.")
125
Φαρμακοκινητικές ιδιότητες περιορ. φάσματος πενικιλινών
Πενικιλλίνη G Πενικιλλίνη V Απορρόφηση από ΓΕΣ (%) 20-30% (pH) 50% Σύνδεση με λευκώματα 45% 60% Ημιπερίοδος ζωής (h) 0.5-1 Απέκκριση με ούρα (%) 80-95 (σωληναρ. έκκριση) 30-50 Κατανομή σε νεφρό, πνεύμονες, ήπαρ, οστά, μύες, πλακούντα, υγρά, χολή. ΕΝΥ 1-5% του ορού.
 20-30% (pH) 50% Σύνδεση με λευκώματα. 45% 60% Ημιπερίοδος ζωής (h) Απέκκριση με ούρα (%) (σωληναρ. έκκριση) Κατανομή. σε νεφρό, πνεύμονες, ήπαρ, οστά, μύες, πλακούντα, υγρά, χολή. ΕΝΥ 1-5% του ορού.")
126
Αντιμικροβικό φάσμα πενικιλινών περ
Αντιμικροβικό φάσμα πενικιλινών περ. φάσματος (penicillin G, penicillin V) Gram-θετικά Streptococcus spp S. aureus (συχνή αντοχή) S. pneumoniae (αντοχή) Enterococcus spp Corynebacterium diphtheriae Listeria monocytogenes Bacillus anthracis Gram-αρνητικά Neisseria spp Moraxella catarrhalis (ανθεκτικά) Αναερόβια Πεπτοστρεπτόκοκκος Πεπτόκοκκος Fusobacterium Bacteroides spp Clostridium spp Σπειροχαίτες Treponema pallidum Borrelia spp Lepstospira spp Spirilium minus Enterococci are facultatively anaerobic, gram-positive cocci that grow in short chains under extreme conditions, i.e., 6.5% NaCl, pH 9.6, temp range from 10-45°C, and in the presence of bile salts. Can be found in soil, water, food, and are significant component of normal colonic flora. Also, in oropharyngeal and vaginal secretions. Relatively low virulence; adheres to extracellular matrix proteins, adheres to urinary tract epithelia, and produces biofilms. Capable of inciting sepsis and septic shock, especially in severely ill pts. Intrinsically resistant to beta-lactams due to inner cell wall penicillin-binding proteins. High level beta-lactam resistance is increasing inE. faecium, but uncommon in E. faecalis. Resistant to TMP-SMX, as organism uses exogenous folate to overcome anti-folate synthesis mechanism. Relatively impermeable to aminoglycosides, adequate drug concentrations may be achieved with addition of a cell-wall agent and result in bactericidal effect at ribosomal target. Ribosomal mutation and decreased aminoglycoside transport confer high-level resistance. Some may remain susceptible to streptomycin. Vancomycin resistant enterococci (VRE): E. faecium (80%) >> E. faecalis (7%) in device-associated infection reported by National Health Safety Network. Plasmid-mediated VanA and VanB gene complexes confer high-level vancomycin resistance. Increased incidence of vancomycin resistant E. faecium attributed to emergence of clonal cluster 17 genogroup. Gram θετικοί κόκκοι Στρεπτόκοκκοι Ομάδας A, B, C, G, bovis, viridans, S. pneumonia (αντοχή στην Ελλάδα: IR/R: %/10-15%) Πεπτοστρεπτόκοκκος και πεπτόκοκκος Εντερόκοκκος (περιορισμένη ευαισθησία). Σταφυλόκοκκοι (β λακταμάση αρνητικοί). Gram αρνητικοί κόκκοι N. meningitidis, N. gonorrhoeae, Moraxella catarrhalis (ανθ. στελέχη) Gram (-) αερόβιοι και αναερόβιοι βάκιλλοι Fusobacterium, Bacteroides spp (όχι B. fragilis), Pasteurella spp. Σπειροχαίτες Treponema palidum, Borrelia spp. (burgdorferi, recurrhentis), Lepstospira spp., Spirilium minus.
 Gram-θετικά. Streptococcus spp. S. aureus (συχνή αντοχή) S. pneumoniae (αντοχή) Enterococcus spp. Corynebacterium diphtheriae. Listeria monocytogenes. Bacillus anthracis. Gram-αρνητικά. Neisseria spp. Moraxella catarrhalis (ανθεκτικά) Αναερόβια. Πεπτοστρεπτόκοκκος. Πεπτόκοκκος. Fusobacterium. Bacteroides spp. Clostridium spp. Σπειροχαίτες. Treponema pallidum. Borrelia spp. Lepstospira spp. Spirilium minus. Enterococci are facultatively anaerobic, gram-positive cocci that grow in short chains under extreme conditions, i.e., 6.5% NaCl, pH 9.6, temp range from 10-45°C, and in the presence of bile salts. Can be found in soil, water, food, and are significant component of normal colonic flora. Also, in oropharyngeal and vaginal secretions. Relatively low virulence; adheres to extracellular matrix proteins, adheres to urinary tract epithelia, and produces biofilms. Capable of inciting sepsis and septic shock, especially in severely ill pts. Intrinsically resistant to beta-lactams due to inner cell wall penicillin-binding proteins. High level beta-lactam resistance is increasing inE. faecium, but uncommon in E. faecalis. Resistant to TMP-SMX, as organism uses exogenous folate to overcome anti-folate synthesis mechanism. Relatively impermeable to aminoglycosides, adequate drug concentrations may be achieved with addition of a cell-wall agent and result in bactericidal effect at ribosomal target. Ribosomal mutation and decreased aminoglycoside transport confer high-level resistance. Some may remain susceptible to streptomycin. Vancomycin resistant enterococci (VRE): E. faecium (80%) >> E. faecalis (7%) in device-associated infection reported by National Health Safety Network. Plasmid-mediated VanA and VanB gene complexes confer high-level vancomycin resistance. Increased incidence of vancomycin resistant E. faecium attributed to emergence of clonal cluster 17 genogroup. Gram θετικοί κόκκοι. Στρεπτόκοκκοι. Ομάδας A, B, C, G, bovis, viridans, S. pneumonia (αντοχή στην Ελλάδα: IR/R: %/10-15%) Πεπτοστρεπτόκοκκος και πεπτόκοκκος. Εντερόκοκκος (περιορισμένη ευαισθησία). Σταφυλόκοκκοι (β λακταμάση αρνητικοί). Gram αρνητικοί κόκκοι. N. meningitidis, N. gonorrhoeae, Moraxella catarrhalis (ανθ. στελέχη) Gram (-) αερόβιοι και αναερόβιοι βάκιλλοι. Fusobacterium, Bacteroides spp (όχι B. fragilis), Pasteurella spp. Σπειροχαίτες. Treponema palidum, Borrelia spp. (burgdorferi, recurrhentis), Lepstospira spp., Spirilium minus.")
127
Ενδείξεις Πενικιλλίνης G
Σύφιλη (τριτοπαθής: νευροσύφιλη) Ενδοκαρδίτιδα (από S. viridans ή εντερόκοκκο) Πνευμονιοκοκκική πνευμονία Μηνιγγιτιδοκοκκική μηνιγγίτιδα Νόσος Lyme Aεριογόνος γάγγραινα Τέτανος Διφθερίτιδα Άνθρακας Λοίμωξη μετά από δήγμα ζώου (P. multocida) Λεπτοσπίρωση Ακτινομύκωση
 Ενδοκαρδίτιδα (από S. viridans ή εντερόκοκκο) Πνευμονιοκοκκική πνευμονία. Μηνιγγιτιδοκοκκική μηνιγγίτιδα. Νόσος Lyme. Aεριογόνος γάγγραινα. Τέτανος. Διφθερίτιδα. Άνθρακας. Λοίμωξη μετά από δήγμα ζώου (P. multocida) Λεπτοσπίρωση. Ακτινομύκωση.")
128
Ενδείξεις βενζαθινικής πενικιλλίνης
Δευτεροπαθής πρόληψη ρευματικού πυρετού 2,4 εκμ. i.u.. i.m. / μήνα Θεραπεία σύφιλης πρωτοπαθής, δευτεροπαθής, πρώιμο λανθάνον στάδιο, στάδιο επώασης: 2,4 εκμ. i.u. εφ’ άπαξ, i.m. όψιμο λανθάνον στάδιο, καλοήθης τριτοπαθής, καρδιαγγειακή σύφιλη: 2,4 εκμ. i.u. i.m. / εβδομάδα (x3).
.")
129
Κλινικές Ενδείξεις Πενικιλλίνης V
Στρεπτοκοκκική φαρυγγoαμυγδαλίτιδα Λοιμώξεις στόματος και περιοδοντικές Συνέχιση ΕΦ θεραπείας με πενικιλλίνη.

130
ΠΕΝΙΚΙΛΙΝΕΣ Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V
* Πενικιλίνες περιορισμένου φάσματος Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V * Ανθεκτικές στην πενικιλινάση πενικιλίνες Μεθικιλλίνη, οξα-, δικλο-, κλοξα- φλουκλοξα, ναφ-κιλλίνη * Πενικιλίνες ευρέος φάσματος Αμινοπενικιλίνες (αμπικιλλίνη, αμοξυκιλίνη) Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη Penicillin G Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections
 Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης. αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ. τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη. Penicillin G. Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections.")
131
ΑΝΘΕΚΤΙΚΕΣ ΣΤΗΝ ΠΕΝΙΚΙΛΛΙΝΑΣΗ Ή ΑΝΤΙΣΤΑΦΥΛΟΚΟΚΚΙΚΕΣ ΠΕΝΙΚΙΛΛΙΝΕΣ
Μεθικιλλίνη (εισήχθη το 1959) Οξακιλλίνη Δικλοξακιλλίνη (Diclocil®) Κλοξακιλλίνη (Orbenin®, Staphyclox®) Φλουκλοξακιλλίνη (Floxapen®, Itaclox®) Nαφκιλλίνη.
 Οξακιλλίνη. Δικλοξακιλλίνη (Diclocil®) Κλοξακιλλίνη (Orbenin®, Staphyclox®) Φλουκλοξακιλλίνη (Floxapen®, Itaclox®) Nαφκιλλίνη.")
132
Φαρμακοκινητικές ιδιότητες αντισταφυλοκ. πενικιλλινών
Μεθικ. Οξακ. Κλοξακ.. Δικλοξ. Φλουκλ. Ναφκιλ. Απορ. από ΓΕΣ (%) - 30 30-50 50 10-20 Σύνδεση με λεύκωμα (%) 35-40 90 95 87-90 Ημιπερ. ζωής (h) 0.5-1 0.5 Αποβολή νεφρική ηπατική Κατανομή Σε νεφρό, πνεύμονες, ήπαρ, οστά, μυς, πλακούντα, υγρά, χολή. ΕΝΥ: 1-5%.
 Σύνδεση με λεύκωμα (%) Ημιπερ. ζωής (h) Αποβολή. νεφρική. ηπατική. Κατανομή. Σε νεφρό, πνεύμονες, ήπαρ, οστά, μυς, πλακούντα, υγρά, χολή. ΕΝΥ: 1-5%.")
133
Δραστικό φάσμα αντισταφυλοκοκκικών πενικιλλινών
Το φάσμα της πενικιλλίνης G λιγότερο δραστικές {1/10 έναντι Gram+ } Σταφυλόκοκκοι (PRSA, αλλά MSSA) Συχνή αντοχή (MRSA) HA- MRSA : 36-60% σε ελληνικά νοσοκομεία CA- MRSA: των σταφ. λοιμώξεων της κοινότητας.
 Συχνή αντοχή (MRSA) HA- MRSA : 36-60% σε ελληνικά νοσοκομεία. CA- MRSA: των σταφ. λοιμώξεων της κοινότητας.")
135
MRSA ΣΕ ΕΛΛΗΝΙΚΑ ΝΟΣΟΚΟΜΕΙΑ (%) (Ιανουάριος- Ιούνιος 2010, WHONET)
 (Ιανουάριος- Ιούνιος 2010, WHONET)")
136
Timeline of Staphylococcal antibiotic resistance
Penicillin-resistance Sporadic MRSA Epidemic MRSA GISA CA-MRSA VRSA 1940 1950 1960 1970 1980 1990 2000 2010 136

137
Ενδείξεις αντισταφυλοκοκκικών πενικιλλινών
Σταφυλοκοκκικές λοιμώξεις (πενικιλλινάση +) Δοθιήνωση Λοιμώξεις δέρματος και μαλακών μορίων Οστεομυελίτιδα Σηπτική αρθρίτιδα Πνευμονία Ενδοκαρδίτιδα Σταφυλοκοκκική σήψη. Για λοιμώξεις από MSSA οι αντισταφυλοκοκκικές πενικιλλίνες και άλλα β-λακταμικά αντιβιοτικά είναι αποτελεσματικότερα των γλυκοπεπτιδίων
 Δοθιήνωση. Λοιμώξεις δέρματος και μαλακών μορίων. Οστεομυελίτιδα. Σηπτική αρθρίτιδα. Πνευμονία. Ενδοκαρδίτιδα. Σταφυλοκοκκική σήψη. Για λοιμώξεις από MSSA οι αντισταφυλοκοκκικές πενικιλλίνες και άλλα β-λακταμικά αντιβιοτικά είναι αποτελεσματικότερα των γλυκοπεπτιδίων.")
138
ΠΕΝΙΚΙΛΙΝΕΣ Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V
* Πενικιλίνες περιορισμένου φάσματος Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V * Ανθεκτικές στην πενικιλινάση πενικιλίνες Μεθικιλλίνη, οξα-, δικλο-, κλοξα- φλουκλοξα, ναφ-κιλλίνη * Πενικιλίνες ευρέος φάσματος Αμινοπενικιλίνες (αμπικιλλίνη, αμοξυκιλίνη) Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη Penicillin G Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections
 Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης. αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ. τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη. Penicillin G. Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections.")
139
Η αμοξυκιλλίνη είναι υδροξυλιωμένο παράγωγο της αμπικιλλίνης
ΑΜΙΝΟΠΕΝΙΚΙΛΛΙΝΕΣ Αμπικιλλίνη Αμοξυκιλλίνη Μπακαμπικιλλίνη Πιβαμπικιλλίνη Επικιλλίνη Ετακιλλίνη. Οι μόνες εν χρήσει σήμερα Υδρολύονται σε αμπικιλίνη Η αμοξυκιλλίνη είναι υδροξυλιωμένο παράγωγο της αμπικιλλίνης

140
Φαρμακοκινητικές ιδιότητες
Αμπικιλλίνη Αμοξυκιλλίνη Απορρόφηση από ΓΕΣ (%) 30-50% (pH) 70-95% Σύνδεση με λευκώματα 20% Ημιπερίοδος (h) 1.3 Αποβολή Νεφρική Μεταβολισμός 15-50% <30% Κατανομή σε νεφρό, πνεύμονες, ήπαρ, oστά, μυς, πλακούντα, υγρά, χολή. ΕΝΥ 1-5% του ορού.
 30-50% (pH) 70-95% Σύνδεση με λευκώματα. 20% Ημιπερίοδος (h) 1.3. Αποβολή. Νεφρική. Μεταβολισμός % <30% Κατανομή. σε νεφρό, πνεύμονες, ήπαρ, oστά, μυς, πλακούντα, υγρά, χολή. ΕΝΥ 1-5% του ορού.")
141
Δραστικό φάσμα αμινοπενικιλλινών
Της πενικιλλίνης G Μικρότερη δραστικότητα έναντι κόκκων (25-50%) Μεγαλύτερη δραστικότητα έναντι λιστέριας και εντερόκοκκου Moraxella catarrhalis (ανθ. στελέχη έως 90%) Haemophilus influenzae (ανθ.στελέχη έως 30%) Εντεροβακτηριακά E. coli, Prοteus mirabilis, Salmonella spp, Shigella spp. (ανθ. στελέχη). Της πενικιλλίνης G Έναντι κόκκων: 1/2-1/4 Έναντι λιστέριας και εντερόκοκκου: > Haemophilus influenzae (ανθ.στελέχη έως 30%) Moraxella catarrhalis (ανθ. στελέχη έως 90%) E. coli, Prοteus mirabilis, Salmonella spp, Shigella spp. (συχνά ανθεκτικά στελέχη).
 Μεγαλύτερη δραστικότητα έναντι λιστέριας και εντερόκοκκου. Moraxella catarrhalis (ανθ. στελέχη έως 90%) Haemophilus influenzae (ανθ.στελέχη έως 30%) Εντεροβακτηριακά. E. coli, Prοteus mirabilis, Salmonella spp, Shigella spp. (ανθ. στελέχη). Της πενικιλλίνης G. Έναντι κόκκων: 1/2-1/4. Έναντι λιστέριας και εντερόκοκκου: > Haemophilus influenzae (ανθ.στελέχη έως 30%) Moraxella catarrhalis (ανθ. στελέχη έως 90%) E. coli, Prοteus mirabilis, Salmonella spp, Shigella spp. (συχνά ανθεκτικά στελέχη).")
142
Ενδείξεις αμινοπενικιλλινών
Λοιμώξεις από εντερόκοκκο (+ αμινογλυκοσίδη) Λιστερίωση Ανεπίπλεκτες ουρολοιμώξεις Λοιμώξεις αναπνευστικού Λοιμώξεις χοληφόρων (σε ευαίσθητα παθογόνα) Σιγκέλλωση (σε ευαίσθητα παθογόνα)
 Λιστερίωση. Ανεπίπλεκτες ουρολοιμώξεις. Λοιμώξεις αναπνευστικού. Λοιμώξεις χοληφόρων (σε ευαίσθητα παθογόνα) Σιγκέλλωση (σε ευαίσθητα παθογόνα)")
143
Ανεπιθύμητες ενέργειες αμινοπενικιλλινών
Παρόμοιες με πενικιλλίνης G Κηλιδοβλατιδώδες εξάνθημα, συχνότερα της πενικιλίνης σε EBV ή λεμφογενή λευχαιμία (90-100%) σε συνδυασμό με αλλοπουρινόλη (14-22%) σε ασθενείς με AIDS Γαστρεντερικές διαταραχές (5-20%).
 σε συνδυασμό με αλλοπουρινόλη (14-22%) σε ασθενείς με AIDS. Γαστρεντερικές διαταραχές (5-20%).")
144
ΠΕΝΙΚΙΛΙΝΕΣ Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V
* Πενικιλίνες περιορισμένου φάσματος Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V * Ανθεκτικές στην πενικιλινάση πενικιλίνες Μεθικιλλίνη, οξα-, δικλο-, κλοξα- φλουκλοξα, ναφ-κιλλίνη * Πενικιλίνες ευρέος φάσματος Αμινοπενικιλίνες (αμπικιλλίνη, αμοξυκιλίνη) Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη Penicillin G Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections
 Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης. αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ. τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη. Penicillin G. Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections.")
145
ΚΑΡΒΟΞΥΛΟΠΕΝΙΚΙΛΛΙΝΕΣ (τρίτης γενεάς πενικιλλίνες)
ΚΑΡΒΟΞΥΛΟΠΕΝΙΚΙΛΛΙΝΕΣ (τρίτης γενεάς πενικιλλίνες) Καρβενικιλλίνη Pyopen®, δεν κυκλοφορεί Τικαρκιλλίνη Neopyopen®, δεν κυκλοφορεί Καρφεκιλλίνη Purapen® , δεν κυκλοφορεί
 Καρβενικιλλίνη. Pyopen®, δεν κυκλοφορεί. Τικαρκιλλίνη. Neopyopen®, δεν κυκλοφορεί. Καρφεκιλλίνη. Purapen® , δεν κυκλοφορεί.")
146
Αντιμικροβιακό φάσμα καρβοξυλοπενικιλλινών
Της αμπικιλλίνης για Gram θετικά Μειωμένη δραστικότητα τικαρκιλίνης έναντι E. faecalis Εντεροβακτηριακά E. coli, Proteus spp., Enterobacter spp Ψευδομονάδα Αναερόβια 50% των στελεχών Β. fragilis.

147
Φαρμακοκινητικές ιδιότητες καρβοξυλοπενικιλλινών
Καρβενικιλ. Τικαρκιλ. Καρφεκιλ. Απορρόφηση από ΓΕΣ (%) όχι καλή Σύνδεση με λευκώματα 50% 20% Ημιπερίοδος (h) 1 1.3 Απέκκριση (σε 8h) Ν (50-90%) Ν (58-93%) Ν (40-50%) Κατανομή σε νεφρό, πνεύμονες, ήπαρ, οστά, μυες, πλακούντα, υγρά, χολή. ΕΝΥ 1-5% του ορού.
 όχι. καλή. Σύνδεση με λευκώματα. 50% 20% Ημιπερίοδος (h) Απέκκριση (σε 8h) Ν (50-90%) Ν (58-93%) Ν (40-50%) Κατανομή. σε νεφρό, πνεύμονες, ήπαρ, οστά, μυες, πλακούντα, υγρά, χολή. ΕΝΥ 1-5% του ορού.")
148
Περιεκτικότητα σκευασμάτων σε Νάτριο
Καρβενικιλλίνη (Pyopen): 4,7 mEq/g Τικαρκιλλίνη (Neopyopen): 5,1 mEq/g Καρφεκιλλίνη (Purapen): 4,7-5,3 mEq/g
: 4,7 mEq/g. Τικαρκιλλίνη (Neopyopen): 5,1 mEq/g. Καρφεκιλλίνη (Purapen): 4,7-5,3 mEq/g.")
149
Πιπερακιλλίνη (Piplril®) Αζλοκιλλίνη (Abrodil®) Μεζλοκιλλίνη
ΑΚΥΛΑΜΙΝΟΠΕΝΙΚΙΛΛΙΝΕΣ ή ΟΥΡΕΪΔΟΠΕΝΙΚΙΛΛΙΝΕΣ (Τετάρτης γενεάς πενικιλλίνες) Πιπερακιλλίνη (Piplril®) Αζλοκιλλίνη (Abrodil®) Μεζλοκιλλίνη Απαλκιλλίνη. Δεν κυκλοφορούν
 Πιπερακιλλίνη (Piplril®) Αζλοκιλλίνη (Abrodil®) Μεζλοκιλλίνη. Απαλκιλλίνη. Δεν κυκλοφορούν.")
150
Φαρμακοκινητικές ιδιότητες
Αζλοκιλίνη Μεζλοκιλίνη Πιπερακιλίνη Απορ. από ΓΕΣ - Σύνδεση με λευκώματα 30% Ημιπερ. (h) 0,6-1,3 Απέκ. με ούρα (% σε 8h) Ν (55-70) Απέκ. στη χολή x15 ορού καλή πολλαπλάσια ορού Κατανομή σε νεφρό, πνεύμονες, ήπαρ, οστά, μυς, πλακούντα, υγρά. ΕΝΥ 1-5%.
 0,6-1,3. Απέκ. με ούρα (% σε 8h) Ν (55-70) Απέκ. στη χολή. x15 ορού. καλή. πολλαπλάσια ορού. Κατανομή. σε νεφρό, πνεύμονες, ήπαρ, οστά, μυς, πλακούντα, υγρά. ΕΝΥ 1-5%.")
151
Αντιμικροβιακό Φάσμα ουρεϊδοπενικιλλινών
Της αμπικιλλίνης Εντεροβακτηριακά (ανθεκτικά στελέχη) Pseudomonas aeruginosa αζλοκιλλίνη & πιπερακιλλίνη: 10x δραστικότερες καρβενικιλλίνης Αναερόβια βακτήρια (B. fragiliς)
 Pseudomonas aeruginosa. αζλοκιλλίνη & πιπερακιλλίνη: 10x δραστικότερες καρβενικιλλίνης. Αναερόβια βακτήρια (B. fragiliς)")
152
Aνεπιθύμητες ενέργειες ουρεϊδοπενικιλλινών
Παρόμοιες των άλλων πενικιλλινών. Παροδική αύξηση ηπατικών ενζύμων Ανατάξιμη ουδετεροπενία Αθροίζονται σε νεφρική ανεπάρκεια Περιεκτικότητα σε Νάτριο Πιπερακιλλίνη: 1,85 mEq/g Αζλοκιλλίνη : 2,17 mEq/g Μεζλοκιλλίνη : 1,85 mEq/g

153
ΑΜΙΔΙΝΟΠΕΝΙΚΙΛΛΙΝΕΣ Μεκιλλινάμη (Seleχid-N®, inj, IM, IV VIAL, 1g) Πιβμεκιλλινάμη (Seleχid® , Film c. tbl, 200 mg).
.")
154
Αντιμικροβιακό Φάσμα αμιδινοπενικιλλινών
Εντεροβακτηριακά: E. coli Proteus mirabilis, P. vulgaris Klebsiella pneumonia Enterobacter Citrobacter Yersinia spp Salnonella spp Shigella spp.

155
Κλινικές ενδείξεις αμιδινοπενικιλλινών
Λοιμώξεις ουροποιητικού Σιγκελλώσεις Λοιμώξεις αναπνευστικού από εντεροβακτηριακά Εμπειρικώς σε συνδυασμό με άλλα β-λακταμικά.

156
ΠΕΝΙΚΙΛΙΝΕΣ Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V
* Πενικιλίνες περιορισμένου φάσματος Πενικιλίνη G, βενζαθινική πενικιλίνη , πενικιλίνη V * Ανθεκτικές στην πενικιλινάση πενικιλίνες Μεθικιλλίνη, οξα-, δικλο-, κλοξα- φλουκλοξα, ναφ-κιλλίνη * Πενικιλίνες ευρέος φάσματος Αμινοπενικιλίνες (αμπικιλλίνη, αμοξυκιλίνη) Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη Penicillin G Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections
 Καρβοξυλοπενικιλίνες (τικαρκιλίνη) Ουρεϊδοπενικιλίνες (πιπερακιλίνη) Αμιδινοπενικιλίνες (μεκιλλινάμη, πιβμεκιλλινάμη) * Πενικιλίνη + αναστολέας β λακταμάσης. αμπικιλίνη / σουλμπακτάμη, αμοξυκιλίνη / κλαβουλανικό οξύ. τικαρκιλίνη + κλαβουλανικό οξύ, πιπερακιλίνη + ταζομπακτάμη. Penicillin G. Still useful for a number of diseases (e.g. meningitis, syphilis) For MSSA infections.")
157
ΑΝΑΣΤΟΛΕΙΣ β-ΛΑΚΤΑΜΑΣΗΣ
Κλαβουλανικό οξύ Σουλμπακτάμη Ταζομπακτάμη Έχουν εγκριθεί για χρήση σε συνδυασμό με β-λακταμικά αντιβιοτικά Διαθέτουν δραστικότητα έναντι Acinetobacter sp.

158
ΠΕΝΙΚΙΛΛΙΝΕΣ + ΑΝΑΣΤΟΛΕΙΣ β-ΛΑΚΤΑΜΑΣΗΣ
Αμοξ. (250 ή 500mg) + Κλ. οξύ (62.5 ή 125mg) (Augmentin®) tbl, susp) Aμοξ. (0.5 ή 1g) + Κλ. οξύ (100 ή 200 mg) (Augmentin® inj) Aμπ. (300 mg) + Σουλμ. (75 mg) (sultamicillin) (Begallin® tbl) Aμπ. (200 mg) + Σουλμ. (50 mg) (Begallin® susp) Aμπ. (1 ή 2g) + Σουλμ. (0,5 ή 1g) (Begallin-P®, inj) Tικαρσ. (3g ή 5g)+ Κλ. οξύ (200mg) (Timentin®) Πιπ. (2 ή 4g)+Ταζ. (250 ή 500 mg) (Tazocin®, inj) Drugs Apr;37(4): Sultamicillin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Friedel HA, Campoli-Richards DM, Goa KL. ADIS Drug Information Services, Auckland, New Zealand. Erratum in: Abstract Sultamicillin is the tosylate salt of the double ester of sulbactam plus ampicillin. Sulbactam is a semisynthetic beta-lactamase inhibitor which, in combination with ampicillin, extends the antibacterial activity of the latter to include some beta-lactamase-producing strains of bacteria that would otherwise be resistant. The combination of sulbactam plus ampicillin for parenteral use has previously been shown to be clinically and bacteriologically effective in a variety of infections. The chemical linkage of sulbactam and ampicillin has now produced an orally effective compound, sultamicillin, with antibacterial activity and clinical efficacy which are similar to those of the parenteral formulation. Sultamicillin has been shown to be clinically effective in non-comparative trials in patients with infections of the respiratory tract, ears, nose and throat, urinary tract, skin and soft tissues, as well as in obstetric and gynaecological infections, and in the treatment of gonorrhoea. In a small number of controlled trials, sultamicillin has shown comparable clinical efficacy to phenoxymethyl penicillin (penicillin V) and to amoxycillin (alone and in combination with clavulanic acid) in the treatment of paediatric streptococcal pharyngitis and acute otitis media, respectively; to cefaclor in the treatment of acute otitis media in adults; and to bacampicillin, cloxacillin and flucloxacillin plus ampicillin in skin and soft tissue infections in adults, children and adult diabetic patients, respectively. Sultamicillin was superior in efficacy to bacampicillin in the treatment of chronic respiratory infections, to cefaclor in the treatment of acute otitis media in adults, and to cefadroxil in the treatment of patients with complicated urinary tract infections. However, in single-dose treatment of uncomplicated gonorrhoea, sultamicillin (1500mg plus probenecid 1g) was inferior to a 2g intramuscular dose of spectinomycin. While in several studies the incidence of diarrhoea associated with sultamicillin was greater than that with comparative antibacterials, sultamicillin-associated diarrhoea was generally mild and transitory, although occasionally severe enough to necessitate discontinuation of treatment. Further studies in larger groups of patients are needed to clarify the therapeutic efficacy and safety of sultamicillin in comparison with other antibacterial regimens, and to determine the optimum single dosage for the treatment of gonorrhoea. Nonetheless, sultamicillin appears to provide a similar pharmacodynamic and pharmacokinetic profile to that of parenteral sulbactam plus ampicillin and, as such, will extend the therapeutic efficacy of ampicillin, with the further advantage of allowing treatment of patients with an oral formulation, thus avoiding the potentially adverse clinical and financial effects of prolonged parenteral therapy
 + Κλ. οξύ (62.5 ή 125mg) (Augmentin®) tbl, susp) Aμοξ. (0.5 ή 1g) + Κλ. οξύ (100 ή 200 mg) (Augmentin® inj) Aμπ. (300 mg) + Σουλμ. (75 mg) (sultamicillin) (Begallin® tbl) Aμπ. (200 mg) + Σουλμ. (50 mg) (Begallin® susp) Aμπ. (1 ή 2g) + Σουλμ. (0,5 ή 1g) (Begallin-P®, inj) Tικαρσ. (3g ή 5g)+ Κλ. οξύ (200mg) (Timentin®) Πιπ. (2 ή 4g)+Ταζ. (250 ή 500 mg) (Tazocin®, inj) Drugs Apr;37(4): Sultamicillin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Friedel HA, Campoli-Richards DM, Goa KL. ADIS Drug Information Services, Auckland, New Zealand. Erratum in: Abstract. Sultamicillin is the tosylate salt of the double ester of sulbactam plus ampicillin. Sulbactam is a semisynthetic beta-lactamase inhibitor which, in combination with ampicillin, extends the antibacterial activity of the latter to include some beta-lactamase-producing strains of bacteria that would otherwise be resistant. The combination of sulbactam plus ampicillin for parenteral use has previously been shown to be clinically and bacteriologically effective in a variety of infections. The chemical linkage of sulbactam and ampicillin has now produced an orally effective compound, sultamicillin, with antibacterial activity and clinical efficacy which are similar to those of the parenteral formulation. Sultamicillin has been shown to be clinically effective in non-comparative trials in patients with infections of the respiratory tract, ears, nose and throat, urinary tract, skin and soft tissues, as well as in obstetric and gynaecological infections, and in the treatment of gonorrhoea. In a small number of controlled trials, sultamicillin has shown comparable clinical efficacy to phenoxymethyl penicillin (penicillin V) and to amoxycillin (alone and in combination with clavulanic acid) in the treatment of paediatric streptococcal pharyngitis and acute otitis media, respectively; to cefaclor in the treatment of acute otitis media in adults; and to bacampicillin, cloxacillin and flucloxacillin plus ampicillin in skin and soft tissue infections in adults, children and adult diabetic patients, respectively. Sultamicillin was superior in efficacy to bacampicillin in the treatment of chronic respiratory infections, to cefaclor in the treatment of acute otitis media in adults, and to cefadroxil in the treatment of patients with complicated urinary tract infections. However, in single-dose treatment of uncomplicated gonorrhoea, sultamicillin (1500mg plus probenecid 1g) was inferior to a 2g intramuscular dose of spectinomycin. While in several studies the incidence of diarrhoea associated with sultamicillin was greater than that with comparative antibacterials, sultamicillin-associated diarrhoea was generally mild and transitory, although occasionally severe enough to necessitate discontinuation of treatment. Further studies in larger groups of patients are needed to clarify the therapeutic efficacy and safety of sultamicillin in comparison with other antibacterial regimens, and to determine the optimum single dosage for the treatment of gonorrhoea. Nonetheless, sultamicillin appears to provide a similar pharmacodynamic and pharmacokinetic profile to that of parenteral sulbactam plus ampicillin and, as such, will extend the therapeutic efficacy of ampicillin, with the further advantage of allowing treatment of patients with an oral formulation, thus avoiding the potentially adverse clinical and financial effects of prolonged parenteral therapy.")
159
Αντιμικροβιακό φάσμα πενικιλλίνης + αναστολέα β-λακταμάσης
Αντιμικροβιακό φάσμα πενικιλλίνης + αναστολέα β-λακταμάσης Της αμπικιλλίνης (+ ανθ. λόγω β-λακταμάσης) Αναερόβια (B. fragilis) Staph. aureus & S. epidermidis (εκτός MRSA) Haemophilus influenzae Moraxella catarrhalis Neisseria gonorrhoeae Εντεροβακτηριακά Escherichia coli, Klebsiela pneumonia, Proteus mirabilis, P. vulgaris Pseudomonas aeruginosa (για ουρεϊδο- & καρβοξυλοπενικιλίνες)
 Αναερόβια (B. fragilis) Staph. aureus & S. epidermidis (εκτός MRSA) Haemophilus influenzae. Moraxella catarrhalis. Neisseria gonorrhoeae. Εντεροβακτηριακά. Escherichia coli, Klebsiela pneumonia, Proteus mirabilis, P. vulgaris. Pseudomonas aeruginosa (για ουρεϊδο- & καρβοξυλοπενικιλίνες)")
160
Ενδείξεις πενικιλ.+αναστ. β-λακταμάσης
Λοιμώξεις αναπνευστικού παραρρινοκολπίτιδα, ωτίτιδα, πνευμονία) Λοιμώξεις ουροποιητικού Δήγματα ανθρώπων & ζώων Γυναικολογικές & ενδοκοιλιακές λοιμώξεις. Λοιμώξεις δέρματος, μαλακών μορίων, οστών.
 Λοιμώξεις ουροποιητικού. Δήγματα ανθρώπων & ζώων. Γυναικολογικές & ενδοκοιλιακές λοιμώξεις. Λοιμώξεις δέρματος, μαλακών μορίων, οστών.")
161
Ανεπιθύμητες ενέργειες πενικιλλίνης + αναστ. β-λακταμάσης
Ανεπιθύμητες ενέργειες πενικιλλίνης + αναστ. β-λακταμάσης Των βασικών ενώσεων (αμπικιλλίνη, τικαρκιλλίνη, πιπερακιλλίνη) Των αναστολέων β-λακταμάσης Γαστρεντερικές διαταραχές κυρίως σε per Os χορήγηση
 Των αναστολέων β-λακταμάσης. Γαστρεντερικές διαταραχές. κυρίως σε per Os χορήγηση.")
162
ΚΕΦΑΛΟΣΠΟΡΙΝΕΣ 1ης γενεάς: 2ης γενεάς: 3ης γενεάς: 4ης γενεάς:
Cefazolin, cefazedone cefalexin, cefadroxil (per Os) 2ης γενεάς: Cefuroxime, cefamandole, cefoxitine, ceforanide, cefaclor, cefprozil, cefuroxime axetile, loracarbef (per Os) 3ης γενεάς: Cefotaxime, Ceftriaxone (Ομάδα 3a) Ceftazidime, cefoperazone (Ομάδα 3b) Cefditoren pivoxil, Cefpodoxime proxetil, Cefixime (per Os) 4ης γενεάς: Cefepime, Cefpirome
 2ης γενεάς: Cefuroxime, cefamandole, cefoxitine, ceforanide, cefaclor, cefprozil, cefuroxime axetile, loracarbef (per Os) 3ης γενεάς: Cefotaxime, Ceftriaxone (Ομάδα 3a) Ceftazidime, cefoperazone (Ομάδα 3b) Cefditoren pivoxil, Cefpodoxime proxetil, Cefixime (per Os) 4ης γενεάς: Cefepime, Cefpirome.")
163
Κατάταξη και δραστικότητα κεφαλοσπορινών
Gram-θετικά (στρεπτόκοκκοι, σταφυλόκοκκοι) Gram-αρνητικά (εντεροβακτηριακά, H. influenzae) 1ης γενεάς +++ + 2ης γενεάς ++ ++ +++ 3ης γενεάς + 4ης γενεάς + +++
 Gram-αρνητικά. (εντεροβακτηριακά, H. influenzae) 1ης γενεάς ης γενεάς ης γενεάς. + 4ης γενεάς")
164
Αδρές διαφορές κεφαλοσπορινών
Δραστικότητα 1ης Γεν. 2ης Γεν. 3ης Γεν. H. influenzae όχι καλή πολύ καλή Gram θετ. κόκκοι (πλην MRSA & εντεροκόκκων) ασταθής Αντοχή σε β λακταμάσες Gram αρνητικών
 ασταθής. Αντοχή σε β λακταμάσες Gram αρνητικών.")
165
Φαρμακοκινητικές ιδιότητες κεφαλοσπορινών
Απεκκρίνονται από νεφρούς (κεφτριαξόνη 50%) Έχουν μικρή ημιπερίοδο ζωής (κεφτριαξόνη 8h) Ικανοποιητική συγκέντρωσή στη χολή κεφαμανδόλη, κεφοξιτίνη, κεφτριαξόνη. Διέρχονται τον αιματοεγκεφαλικό φραγμό κεφουροξίμη, κεφτριαξόνη, κεφοταξίμη, κεφεπίμη.
 Έχουν μικρή ημιπερίοδο ζωής (κεφτριαξόνη 8h) Ικανοποιητική συγκέντρωσή στη χολή. κεφαμανδόλη, κεφοξιτίνη, κεφτριαξόνη. Διέρχονται τον αιματοεγκεφαλικό φραγμό. κεφουροξίμη, κεφτριαξόνη, κεφοταξίμη, κεφεπίμη.")
166
Φάσμα κεφαλοσπορινών πρώτης γενεάς
Gram θετικοί κόκκοι Σταφυλόκοκκοι (πλην MRSA) Στρεπτόκοκκοι (όχι εντερόκοκκοι) Αναερόβιοι στρεπτόκοκκοι Gram αρνητικοί κόκκοι Γονόκοκκος, μηνιγγιτιδόκοκκος (μικρή δραστικότα) Gram θετικοί βάκιλλοι: Κορυνοβακτηρίδιο διφθερίτιδας Ακτινομύκητες, Κλωστηρίδια Gram αρνητικοί βάκιλλοι: (ανθεκτικά στελέχη) E. coli, K. pneumonia, P. mirabilis Salmonella, Sighella sp.
 Στρεπτόκοκκοι (όχι εντερόκοκκοι) Αναερόβιοι στρεπτόκοκκοι. Gram αρνητικοί κόκκοι. Γονόκοκκος, μηνιγγιτιδόκοκκος (μικρή δραστικότα) Gram θετικοί βάκιλλοι: Κορυνοβακτηρίδιο διφθερίτιδας. Ακτινομύκητες, Κλωστηρίδια. Gram αρνητικοί βάκιλλοι: (ανθεκτικά στελέχη) E. coli, K. pneumonia, P. mirabilis. Salmonella, Sighella sp.")
167
ΚΕΦΑΛΟΣΠΟΡΙΝΕΣ ΔΕΥΤΕΡΑΣ ΓΕΝΕΑΣ
Χορηγούμενες παρεντερικώς Κεφαμανδόλη (Mandokef®) Κεφουροξίμη (Zinacef®, κ.ά. Κεφοξιτίνη (Mefoxil®) Κεφορανίδη (Radacef®) Κεφονισίδη Κεφοτετάνη Χορηγούμενες από του στόματος Κεφακλόρη (Ceclor®) Cefuroxim axetyl (Zinadol®) Κεφπροζίλη (Procef ®) Λορακαμπέφη (Lorbef®). Κεφατριζίνη (Cetrizine®,)
 Κεφουροξίμη (Zinacef®, κ.ά. Κεφοξιτίνη (Mefoxil®) Κεφορανίδη (Radacef®) Κεφονισίδη. Κεφοτετάνη. Χορηγούμενες από του στόματος. Κεφακλόρη (Ceclor®) Cefuroxim axetyl (Zinadol®) Κεφπροζίλη (Procef ®) Λορακαμπέφη (Lorbef®). Κεφατριζίνη (Cetrizine®,)")
168
T/ Απορρόφ Σύνδ με λευκ

169
Αντιμικροβιακό φάσμα κεφαλοσπορινών βΓ
Το φάσμα των κεφαλοσπορινών αΓ: Έναντι Gram θετ. κόκκων λιγότερο δραστικές από αΓ Έναντι γονοκόκκου (κεφουροξίμη, κεφοξιτίνη) δραστικότερες Έναντι Gram αρν. βακίλλων (H. influenzae, εντεροβακτηριακά) Έναντι αναερόβιων όπως αΓ κεφοξιτίνη: δραστική έναντι B. fragilis (20% ανθεκτικά στελέχη).
 δραστικότερες. Έναντι Gram αρν. βακίλλων (H. influenzae, εντεροβακτηριακά) Έναντι αναερόβιων όπως αΓ. κεφοξιτίνη: δραστική έναντι B. fragilis (20% ανθεκτικά στελέχη).")
170
Ενδείξεις κεφαλοσπορινών βΓ
Λοιμώξεις αναπνευστικού Ανεπίπλεκτες λοιμώξεις ουροποιητικού Λοιμώξεις δέρματος και μαλακών μορίων Μεικτές αναερόβιες λοιμώξεις περιτονίτιδα, εκκολπωματίτιδα, γυναικ. λοιμώξεις ?. Λοιμώξεις από γονόκοκκο κεφουροξίμη ή κεφοξιτίνη. Χημειοπροφύλαξη στη χειρουργική κεφορανίδη, κεφοξιτίνη και άλλες.

171
ΚΕΦΑΛΟΣΠΟΡΙΝΕΣ TΡΙΤΗΣ ΓΕΝΕΑΣ
Χορηγούμενες παρεντερικώς Kεφοταξίμη (Claforan®) Κεφτριαξόνη (Rocephin®) Κεφταζιδίμη (Ftazidim®, Solvetan®) Κεφοπεραζόνη Χορηγούμενες από το στόμα Cefditoren pivoxil (Spectracef®) Κεφιξίμη, Cefixime (Ceftoral®) Κεφποδοξίμη, Cefpodoxime proxetil (Orelox®) Κεφεταμέτη, Cefetamete pivoxil (Clobocef®) Κεφτιμπουτίνη, Ceftibutine (Ceadex®).
 Κεφτριαξόνη (Rocephin®) Κεφταζιδίμη (Ftazidim®, Solvetan®) Κεφοπεραζόνη. Χορηγούμενες από το στόμα. Cefditoren pivoxil (Spectracef®) Κεφιξίμη, Cefixime (Ceftoral®) Κεφποδοξίμη, Cefpodoxime proxetil (Orelox®) Κεφεταμέτη, Cefetamete pivoxil (Clobocef®) Κεφτιμπουτίνη, Ceftibutine (Ceadex®).")
172
Φάσμα κεφαλοσπορινών γΓ
Το φάσμα των βΓ με τις εξής διαφορές: Έναντι Gram θετ. κόκκων: < λιγότερο δραστικές Έναντι Gram αρν. βακτηρίων: > δραστικότερες H. influenzae E. coli, K. pneumonia, P. mirabilis, P. vulgaris, Enterobacter spp., Serratia spp., Citrobacter spp, Salmonella sp. N. meningitidis, N. gonorhoae. Pseudomonas aeruginosa (μόνο cefoperazone, >ceftazidime)
")
173
Ενδείξεις 3ης γενεάς κεφαλοσπορινών και κεφιπίμης
Οξεία μηνιγγίτιδα Πνευμονία Εμπύρετος ουδετεροπενία Βαριές λοιμώξεις ουροποιητικού ή χοληφόρων Λοιμώξεις από ψευδομονάδα Γονόρροια Νοσοκομειακές λοιμώξεις.

174
ΑΝΕΠΙΘΥΜΗΤΕΣ ΕΝΕΡΓΕΙΕΣ ΚΕΦΑΛΟΣΠΟΡΙΝΩΝ 3ης Γεν.
Ψευδοχολολιθίαση (χολική λάσπη, κυρίως η κεφτριαξόνη) Φλεβίτιδα στο σημείο έγχυσης Αλλεργικές αντιδράσεις Φαρμακευτικός πυρετός Θετική Coombs Διάμεση νεφρίτιδα Διάρροια, κολίτις από C.difficile Σπασμοί και άλλες διαταραχές από ΚΝΣ (σπάνια) Ουδετεροπενία και θρομβοπενία Ηπατίτιδα Αιμολυτική αναιμία Χολοκυστίτιδα
 Φλεβίτιδα στο σημείο έγχυσης. Αλλεργικές αντιδράσεις. Φαρμακευτικός πυρετός. Θετική Coombs. Διάμεση νεφρίτιδα. Διάρροια, κολίτις από C.difficile. Σπασμοί και άλλες διαταραχές από ΚΝΣ (σπάνια) Ουδετεροπενία και θρομβοπενία. Ηπατίτιδα. Αιμολυτική αναιμία. Χολοκυστίτιδα.")
175
Φάσμα κεφαλοσπορινών 4ης γενεάς (cefepime, iv).
Gram-θετ. S. pyogenes, S. viridans, S. pneumoniae. S. aureus (μέτρια δραστικότητα) Gram-αρν. Enterobacteriaceae - E. coli, K. pneumoniae, Proteus spp. κ.ά. H. influenzae, Neisseria spp. Pseudomonas aeruginosa.
 Gram-αρν. Enterobacteriaceae. - E. coli, K. pneumoniae, Proteus spp. κ.ά. H. influenzae, Neisseria spp. Pseudomonas aeruginosa.")
176
ΚΛΙΝΙΚΕΣ ΕΝΔΕΙΞΕΙΣ ΚΕΦΕΠΙΜΗΣ
Πνευμονία από S. pneumoniae, P. aeruginosa, K. pneumoniae, Enterobacter sp. Εμπύρετος ουδετεροπενία (εμπειρική θεραπεία) Επιπλεγμένες και μη λοιμώξεις ουροποιητικού Μη επιπλεγμ. λοιμώξεις δέρματος & μαλακών μορίων από MSSA ή S. pyogenes. Επιπλεγμ. ενδοκοιλ. λοιμώξεις (με metronidazole) Άλλες (μη εγκεκριμένες) Οστεομυελίτιδα, διαβητικό πόδι, κοιλ. λοιμώξεις, λοίμωξη προσθ. άρθρωσης Ιδιαίτερη ανεπιθύμητη ενέργεια: ουδετεροπενία
 Επιπλεγμένες και μη λοιμώξεις ουροποιητικού. Μη επιπλεγμ. λοιμώξεις δέρματος & μαλακών μορίων από MSSA ή S. pyogenes. Επιπλεγμ. ενδοκοιλ. λοιμώξεις (με metronidazole) Άλλες (μη εγκεκριμένες) Οστεομυελίτιδα, διαβητικό πόδι, κοιλ. λοιμώξεις, λοίμωξη προσθ. άρθρωσης. Ιδιαίτερη ανεπιθύμητη ενέργεια: ουδετεροπενία.")
177
Imipenem/cilastin (Primaxin®)
KAΡΒAΠΕΝΕΜΕΣ Imipenem/cilastin (Primaxin®) Meropenem (Meronem®) Ertapenem (Invanz®) Doripenem (Doribax®)
 Meropenem (Meronem®) Ertapenem (Invanz®) Doripenem (Doribax®)")
178
Αντιμικροβιακό φάσμα καρβαπενεμών
Gram θετικά Gram αρνητικά Αναερόβια βακτήρια Βακτήρια που δεν περιλαμβάνονται στο φάσμα Burkholderia cepacia & Stenotrophomonas maltophilia MRSA, Enterococcus faecium Ανθεκτικά στελέχη ή υποδεέστερη δράση Clostridia difficile & διφθεριοειδή E. faecalis & Listeria monocytogenes H. influenzae, N. meningitidis, N. gonorrhea.

179
Αντιμικροβιακό Φάσμα καρβαπενεμών
Αντιβιοτικό Strep spp. & MSSA Entero-bacteriaeae P. aeruginosa Αναερόβια Imipenem + Meropenem Ertapenem - Doripenem

180
Καρβαπενέμες – διαφορές
Imipenem Gram-θετ., Gram-αρν., (+ ESBL), P. aeruginosa, αναερόβια Δοσολογία: 0,5-1 g /6-8 h Meropenem Περίπου ίδια δραστικότητα (> σε ορισμένα Gram αρν., < σε Gram θετ.) Προκαλεί σπανιότερα σπασμούς, μπορεί να χορηγηθεί για λοιμώξεις ΚΝΣ Δοσολογία: 1-2 g /8 h Doripenem Δραστικότερη έναντι P. aeruginosa Ενδείξεις: Νοσ. πνευμονία, επιπλ. λοιμώξεις ουροποιητικού και ενδοκοιλιακές Δοσολογία: 500 mg/8h iv σε 1-3 ώρες (Cl cr mg/8h. Cl cr < mg/12h) Ertapenem Μη δραστικό έναντι Pseudomonas & Acinetobacter δραστικότητα έναντι ανθ. σε πενικιλ. πνευμονιόκ. (PRP) και E. faecalis. Δοσολογία: 1 g / 24 h
, P. aeruginosa, αναερόβια. Δοσολογία: 0,5-1 g /6-8 h. Meropenem. Περίπου ίδια δραστικότητα (> σε ορισμένα Gram αρν., < σε Gram θετ.) Προκαλεί σπανιότερα σπασμούς, μπορεί να χορηγηθεί για λοιμώξεις ΚΝΣ. Δοσολογία: 1-2 g /8 h. Doripenem. Δραστικότερη έναντι P. aeruginosa. Ενδείξεις: Νοσ. πνευμονία, επιπλ. λοιμώξεις ουροποιητικού και ενδοκοιλιακές. Δοσολογία: 500 mg/8h iv σε 1-3 ώρες (Cl cr mg/8h. Cl cr < mg/12h) Ertapenem. Μη δραστικό έναντι Pseudomonas & Acinetobacter. δραστικότητα έναντι ανθ. σε πενικιλ. πνευμονιόκ. (PRP) και E. faecalis. Δοσολογία: 1 g / 24 h.")
181
Κύριες κλινικές ενδείξεις ιμιπενέμης ή μεροπενέμης
Ενδοκοιλιακές λοιμώξεις (μικτές) Εμπύρετη ουδετεροπενία Νοσοκομειακές λοιμώξεις (πνευμονία, κά) Παγκρεατικό απόστημα Σήψη από διάφορες εστίες Λοιμώξεις από πολυανθεκτικά βακτήρια Λοιμώξεις από ψευδομονάδα
 Εμπύρετη ουδετεροπενία. Νοσοκομειακές λοιμώξεις (πνευμονία, κά) Παγκρεατικό απόστημα. Σήψη από διάφορες εστίες. Λοιμώξεις από πολυανθεκτικά βακτήρια. Λοιμώξεις από ψευδομονάδα.")
182
Ανεπιθύμητες ενέργειες καρβαπενεμών
Οι των β λακταμικών αντιβιοτικών Γαστρεντερικές διαταραχές (1-2%) Ψευδομεμβρανώδης κολίτιδα (0.1%) Σπασμοί (imipenem %, meropenem σπανιότερα) Επιλοίμωξη από εντερόκοκκο
 Ψευδομεμβρανώδης κολίτιδα (0.1%) Σπασμοί (imipenem %, meropenem σπανιότερα) Επιλοίμωξη από εντερόκοκκο.")
183
Μπορεί να δοθεί σε περίπτωση αλλεργίας στα β λακταμικά.
Μονοβακτάμες Aztreonam (Azactam®) Φάσμα N. meningitidis & gonorrhea H. influenzae Εντεροβακτηριακά Pseudomonas aeruginosa Ανθεκτικά Burkholderia cepacia Stenotrophomonas maltophilia Acinetobacter Μη δραστική Αναερόβια Gram θετικά Μπορεί να δοθεί σε περίπτωση αλλεργίας στα β λακταμικά.
 Φάσμα. N. meningitidis & gonorrhea. H. influenzae. Εντεροβακτηριακά. Pseudomonas aeruginosa. Ανθεκτικά. Burkholderia cepacia. Stenotrophomonas maltophilia. Acinetobacter. Μη δραστική. Αναερόβια. Gram θετικά. Μπορεί να δοθεί σε περίπτωση αλλεργίας στα β λακταμικά.")
184
Κλινικές ενδείξεις αζτρεονάμης
Οι των κεφαλοσπορινών γΓ (σε αλλεργία) Νοσοκομειακές λοιμώξεις από Gram αρνητικά Λοιμώξεις δέρματος, μυοσκελετικού από Gram αρν. Εμπύρετη ουδετεροπενία (+βανκομυκίνη) Ενδοκοιλιακές ή γυναικολογικές λοιμώξεις (+κλινδαμυκίνη ή μετρονιδαζόλη) Λοιμώξεις από ψευδομονάδα.
 Νοσοκομειακές λοιμώξεις από Gram αρνητικά. Λοιμώξεις δέρματος, μυοσκελετικού από Gram αρν. Εμπύρετη ουδετεροπενία (+βανκομυκίνη) Ενδοκοιλιακές ή γυναικολογικές λοιμώξεις (+κλινδαμυκίνη ή μετρονιδαζόλη) Λοιμώξεις από ψευδομονάδα.")
185
Μέγιστη δοσολογία 2g / 6-8 h
ΧΡΗΣΗ ΑΖΤΡΕΟΝΑΜΗΣ Χορηγείται iv και im t/2 1.7 h (6 h σε ανεφρικούς) Χορήγηση ανά 6-8 h Μέγιστη δοσολογία 2g / 6-8 h
 Χορήγηση ανά 6-8 h. Μέγιστη δοσολογία 2g / 6-8 h.")
186
Κινολόνες Τρίτης γενεάς Τέταρτης γενεάς Παλαιότερες (πρώτης γενεάς)
Nalidic acid, Oxolinic acid, Cinoxacin, Pipemid acid Δεύτερης γενεάς (φθοριοκινολόνες) Norfloxacin, Ofloxacin, Ciprofloxacin, Pefloxacin, Enoxacin Τρίτης γενεάς Levofloxacin Τέταρτης γενεάς Moxifloxacin, Gatifloxacin, Trovafloxacin (ηπατοτοξική)
 Norfloxacin, Ofloxacin, Ciprofloxacin, Pefloxacin, Enoxacin. Τρίτης γενεάς. Levofloxacin Τέταρτης γενεάς. Moxifloxacin, Gatifloxacin, Trovafloxacin (ηπατοτοξική)")
187
Φθοριοκινολόνες Ομάδα 1 (για λοιμώξεις ουροποιητικού)
Norfloxacin, Pefloxacin Ομάδα 2 (για συστηματικές λοιμώξεις) Enoxacin, Fleroxacin, Lomefloxacin, Ofloxacin, Ciprofloxacin Ομάδα 3 (βελτιωμένη δραστικότητα για Gram θετ και άτυπα) Levofloxacin Ομάδα 4 (βελτι. δραστικότητα για Gram θετ., άτυπα και αναερόβια) Moxifloxacin, Gatifloxacin * Listed according to increasing in-vitro activity (minimum inhibitory concentration) against indicative pathogens. ** In France and other countries, pefloxacin is also available for systemic use *** Investigated in acute exacerbations of chronic bronchitis, UTIs, gonorrhoea and gastrointestinal infections. Group 1 fluoroquinolones The indications for group 1 fluoroquinolones is limited to UTIs in some countries, e.g. Germany. In France and some other countries, pefloxacin is also used for systemic oral and parenteral use. Norfloxacin is not available as parenteral antibiotic. Group 2 fluoroquinolones Group 2 fluoroquinolones includes fluoroquinolones for systemic use with a broad spectrum of indications. These include infections of the urinary tract, respiratory tract, skin and soft tissues, bones and joints, as well as systemic infections and even sepsis. Group 2 fluoroquinolones exhibit good activity against enterobacteria and H. influenzae with less activity against staphylococci, pneumococci and enterococci and ‘atypical’ pathogens, e.g. Chlamydia, Legionella and Mycoplasma. Their activity against Ps. aeruginosa varies, with ciprofloxacin being most active in vitro. In addition, ciprofloxacin, ofloxacin and fleroxacin are also available for parenteral use. Group 3 fluoroquinolones The main difference in the spectrums of activity of group 3 fluoroquinolones (levofloxacin) and of group 4 fluoroquinolones (gatifloxacin, moxifloxacin) is that group 3 fluoroquinolones have a higher intrinsic activity against Gram-positive pathogens, such as staphylococci, streptococci, pneumococci and enterococci. However, group 3 and group 4 fluoroquinolones have comparable activity against Gram-negative pathogens. In addition, they have improved activity against the so-called ‘atypical’ pathogens, such as Chlamydia, Mycoplasma and Legionella spp. In addition, group 4 fluoroquinolones have improved anti-anaerobic activity. The only group 3 fluoroquinolone available for parenteral use is levofloxacin, the left enantiomer of the ofloxacin racemate. The main indications for levofloxacin are respiratory tract infections, and, due to its high renal elimination rate, UTIs, as well as skin and soft-tissue infections. Among group 4 fluoroquinolones, gatifloxacin (not on the market in Europe), moxifloxacin and trovafloxacin have been licensed. However, in June 1999, trovafloxacin was taken off the market because of severe side effects. Thus, so far, no parenteral fluoroquinolone of this group has been made available. Apart from respiratory tract infections, these broad-spectrum fluoroquinolones are appropriate for the treatment of skin and soft-tissue infections, of intra-abdominal infections, and of the oral treatment of gynaecological infections. However, final judgement of their position in the treatment of these diseases is not yet possible. Gatifloxacin has the highest renal excretion (about 84%) after oral administration. It is therefore also the most suitable for the treatment of uncomplicated and complicated UTI. The urinary excretion of moxifloxacin after oral administration is only in the range of about 20%. Naber KG, Adam D, and an expert group of the Paul Ehrlich Society for Chemotherapy. Chemotherapie Journal 1998;7:
 Enoxacin, Fleroxacin, Lomefloxacin, Ofloxacin, Ciprofloxacin. Ομάδα 3 (βελτιωμένη δραστικότητα για Gram θετ και άτυπα) Levofloxacin Ομάδα 4 (βελτι. δραστικότητα για Gram θετ., άτυπα και αναερόβια) Moxifloxacin, Gatifloxacin. * Listed according to increasing in-vitro activity (minimum inhibitory concentration) against indicative pathogens. ** In France and other countries, pefloxacin is also available for systemic use. *** Investigated in acute exacerbations of chronic bronchitis, UTIs, gonorrhoea and gastrointestinal infections Group 1 fluoroquinolones. The indications for group 1 fluoroquinolones is limited to UTIs in some countries, e.g. Germany. In France and. some other countries, pefloxacin is also used for systemic oral and parenteral use. Norfloxacin is not available. as parenteral antibiotic Group 2 fluoroquinolones. Group 2 fluoroquinolones includes fluoroquinolones for systemic use with a broad spectrum of indications. These include infections of the urinary tract, respiratory tract, skin and soft tissues, bones and joints, as well as. systemic infections and even sepsis. Group 2 fluoroquinolones exhibit good activity against enterobacteria and. H. influenzae with less activity against staphylococci, pneumococci and enterococci and ‘atypical’ pathogens, e.g. Chlamydia, Legionella and Mycoplasma. Their activity against Ps. aeruginosa varies, with ciprofloxacin. being most active in vitro. In addition, ciprofloxacin, ofloxacin and fleroxacin are also available for parenteral. use Group 3 fluoroquinolones. The main difference in the spectrums of activity of group 3 fluoroquinolones (levofloxacin) and of group 4. fluoroquinolones (gatifloxacin, moxifloxacin) is that group 3 fluoroquinolones have a higher intrinsic activity. against Gram-positive pathogens, such as staphylococci, streptococci, pneumococci and enterococci. However, group 3 and group 4 fluoroquinolones have comparable activity against Gram-negative pathogens. In addition, they have improved activity against the so-called ‘atypical’ pathogens, such as Chlamydia, Mycoplasma and Legionella spp. In addition, group 4 fluoroquinolones have improved anti-anaerobic activity. The only group 3 fluoroquinolone available for parenteral use is levofloxacin, the left enantiomer of the. ofloxacin racemate. The main indications for levofloxacin are respiratory tract infections, and, due to its high. renal elimination rate, UTIs, as well as skin and soft-tissue infections. Among group 4 fluoroquinolones, gatifloxacin (not on the market in Europe), moxifloxacin and. trovafloxacin have been licensed. However, in June 1999, trovafloxacin was taken off the market because of. severe side effects. Thus, so far, no parenteral fluoroquinolone of this group has been made available. Apart from respiratory tract infections, these broad-spectrum fluoroquinolones are appropriate for. the treatment of skin and soft-tissue infections, of intra-abdominal infections, and of the oral treatment of. gynaecological infections. However, final judgement of their position in the treatment of these diseases is not. yet possible. Gatifloxacin has the highest renal excretion (about 84%) after oral administration. It is therefore. also the most suitable for the treatment of uncomplicated and complicated UTI. The urinary excretion of. moxifloxacin after oral administration is only in the range of about 20%. Naber KG, Adam D, and an expert group of the Paul Ehrlich Society for Chemotherapy. Chemotherapie Journal 1998;7:")
188
Αντιμικροβιακό φάσμα νεότερων κινολονών
Streptococcus, Staphylococcus sp Εντεροβακτηριακά Pseudomonas aeruginosa Neisseria meningitidis, N. gonorrhea Moraxella catarrhalis, Haemophilus influenzae Listeria monocytogenes Legionella spp., Χλαμύδια, Μυκόπλασμα Μυκοβακτηρίδια P. aeruginosa: Cipro> Oflo, Levo. S. pneumoniae Moxi> Levo> Cipro Άτυπα Moxi, Levo> Cipro >Oflo Αναερόβια Moxifloxacin

189
AUC/MIC φθοριοκινολονών έναντι S. pneumoniae
50 100 150 200 250 300 350 400 (188–377) (68–298) (65–212) AUC/ MIC If we anticipate that an AUC/MIC somewhere in the range of is an important parameter to maximize clinical efficacy, we can look at a range of anticipated exposures. That is, to look at a range of fluoroquinolones in the context of in vitro susceptibility and with respect to pharmacodynamic profile, we can determine the best and worse case scenarios in patients. We can determine “blocks” of exposure. For compounds such as ciprofloxacin, we see that the exposure is less than adequate. Levofloxacin appears to have a number of patient exposures, which are below what we think to be minimally effective. Newer compounds that have improved microbiologic activity seem to have higher AUC/MIC exposure (gatifloxacin/moxifloxacin) and appear to have levels of exposure that are above what we think is minimally effective. Does this make a difference? (20–44) Ciprofloxacin Levofloxacin Gatifloxacin Moxifloxacin 750 mg 750 mg 400 mg 400 mg Grant & Nicolau Antibiotic for Clinicians 1999;3(Suppl 1):21-28
 (68–298) (65–212) AUC/ MIC. If we anticipate that an AUC/MIC somewhere in the range of is an important parameter to maximize clinical efficacy, we can look at a range of anticipated exposures. That is, to look at a range of fluoroquinolones in the context of in vitro susceptibility and with respect to pharmacodynamic profile, we can determine the best and worse case scenarios in patients. We can determine blocks of exposure. For compounds such as ciprofloxacin, we see that the exposure is less than adequate. Levofloxacin appears to have a number of patient exposures, which are below what we think to be minimally effective. Newer compounds that have improved microbiologic activity seem to have higher AUC/MIC exposure (gatifloxacin/moxifloxacin) and appear to have levels of exposure that are above what we think is minimally effective. Does this make a difference (20–44) Ciprofloxacin. Levofloxacin. Gatifloxacin. Moxifloxacin. 750 mg. 750 mg. 400 mg. 400 mg. Grant & Nicolau Antibiotic for Clinicians 1999;3(Suppl 1):")
190
Levofloxacin Concentrations in Epithelial Lining Fluid
ELF Concentration at Steady State (mg/L) Levofloxacin MIC90 E. coli (0.12) K. pneumoniae (0.12) S. aureus MS (0.5) This graph shows the concentration of drug measured in the epithelial lining fluid after 24 hours of a steady-state oral dose. High-dose levofloxacin maintains a concentration above the MIC value for most potential pathogens even after 24 hours. Time (Hours) Gotfried MH, et al. Chest. 2001;119:
 Levofloxacin MIC90. E. coli (0.12) K. pneumoniae (0.12) S. aureus MS (0.5) This graph shows the concentration of drug measured in the epithelial lining fluid after 24 hours of a steady-state oral dose. High-dose levofloxacin maintains a concentration above the MIC value for most potential pathogens even after 24 hours. Time (Hours) Gotfried MH, et al. Chest. 2001;119:")
191
ΚΙΝΟΛΟΝΕΣ - ΦΑΡΜΑΚΟΚΙΝΗΤΙΚΗ
Σιπροφλο-ξασίνη Οφλο-ξασίνη Λεβο-φλοξασίνη Μοξιφλοξασίνη Απορρόφηση (%) 70-80 98 99 95 Σύνδ. με λευκ. (%) 20-40 32 24-38 20-30 T/2 ( h) 4 4-5 6-8 9,5 Αποβολή 60% νεφρ., 30% ηπατ. 70% νεφρ. 85% νεφρ. 20% νεφρ.
 Σύνδ. με λευκ. (%) T/2 ( h) ,5. Αποβολή. 60% νεφρ., 30% ηπατ. 70% νεφρ. 85% νεφρ. 20% νεφρ.")
192
Συγκέντρωση νεότερων κινολονών στα κυψελιδικά μακροφάγα
Levofloxacin: 18 x ορού Moxifloxacin: 21 x ορού Gatifloxacin: 26 x ορού

193
Ανεπιθύμητες ενέργειες φθοριοκινολονών
Από ΓΕΣ (3-17%) Ναυτία, έμετος, ανορεξία, κοιλιακό άλγος, διάρροια, ψευδομεμβρανώδης κολίτιδα Αλλεργικές αντιδράσεις (0,4-2%) Εξάνθημα, αγγειίτιδα, πυρετός, αγγειοοίδημα, Shock, φωτοδερματίτιδα Από ΚΝΣ (0,9-11%) Κεφαλαλγία, ζάλη, ανησυχία, συγχ. κατάσταση, αϋπνία, ψευδαισθήσεις, ψύχωση, τρόμος, σπασμοί Διαταραχές όρασης, γεύσης, όσφρησης και ακοής.
 Ναυτία, έμετος, ανορεξία, κοιλιακό άλγος, διάρροια, ψευδομεμβρανώδης κολίτιδα. Αλλεργικές αντιδράσεις (0,4-2%) Εξάνθημα, αγγειίτιδα, πυρετός, αγγειοοίδημα, Shock, φωτοδερματίτιδα. Από ΚΝΣ (0,9-11%) Κεφαλαλγία, ζάλη, ανησυχία, συγχ. κατάσταση, αϋπνία, ψευδαισθήσεις, ψύχωση, τρόμος, σπασμοί. Διαταραχές όρασης, γεύσης, όσφρησης και ακοής.")
194
Ανεπιθύμητες ενέργειες φθοριοκινολονών
Από καρδιαγγειακό: Παράταση QT – κοιλιακή ταχυκαρδία (σπάνια) Από αρθρώσεις: Αρθραλγίες (1%) Mη αναστρ. βλάβη χόνδρου μεγ. αρθρώσεων σε νεαρά πειραματόζωα Από ουροποιητικό ( %) Κρυσταλλουρία, αιματουρία, oξ. διάμεση νεφρίτιδα (ΟΝΑ;, ΧΝΑ;) Αιματολογικές και άλλες διαταραχές: Ηωσινοφιλία, ουδετεροπενία, θρομβοπενία, αναιμία Τραναμινασαιμία και άλλες διαταραχές ηπατ. λειτουργίας Headache, dizziness, nausea, lightheadedness Limit use in pregnancy, nursing mothers, and children < 18. Drug interactions: may increase levels of theophylline, warfarin, caffeine and cyclosporine. Absorption decreased when taken with cations. Arthralgias - 1%.
 Από αρθρώσεις: Αρθραλγίες (1%) Mη αναστρ. βλάβη χόνδρου μεγ. αρθρώσεων σε νεαρά πειραματόζωα. Από ουροποιητικό ( %) Κρυσταλλουρία, αιματουρία, oξ. διάμεση νεφρίτιδα (ΟΝΑ;, ΧΝΑ;) Αιματολογικές και άλλες διαταραχές: Ηωσινοφιλία, ουδετεροπενία, θρομβοπενία, αναιμία. Τραναμινασαιμία και άλλες διαταραχές ηπατ. λειτουργίας. Headache, dizziness, nausea, lightheadedness. Limit use in pregnancy, nursing mothers, and children < 18. Drug interactions: may increase levels of theophylline, warfarin, caffeine and cyclosporine. Absorption decreased when taken with cations. Arthralgias - 1%.")
195
Αντενδείξεις θεραπείας με κινολόνες
Ιστορικό υπερευαισθησίας Ιστορικό τενοντίτιδας ή ρήξης τενόντων Βλάβη ΚΝΣ ή ιστορικό σπασμών Έλλειψη G-6-PD; (με προσοχή) Ηλεκτρολυτικές διαταραχές (υποκαλιαιμία) Καρδιολογικά προβλήματα (μοξιφλοξασίνη) Παράταση QTc, καρδιακή ανεπάρκεια με μειωμένο κλάσμα εξώθησης, κλινικά σημαντική βραδυκαρδία, ιστορικό αρρυθμίας Ηλικία < 18 ετών, κύηση και γαλουχία lthough fluoroquinolones are highly effective and safe therapies for adults, they have not been widely prescribed for pediatric patients principally because of laboratory studies demonstrating arthropathy in immature animals following exposure to fluoroquinolones (3, 6, 7, 27). A rather large body of information regarding fluoroquinolone use in children and adolescents for a variety of conditions has accumulated, mainly due to compassionate use in certain pediatric patient populations (1, 3, 14, 27, 29). There has been no report in the scientific literature of a definitive fluoroquinolone-associated case of arthropathy in a child despite several radiological and magnetic resonance imaging investigations (3, 8, 14, 22). Retrospective reviews and prospective studies support the view that fluoroquinolone-associated toxicity unique to children may not occur or occurs at frequencies that are extremely low (1, 3, 14, 23, 27,29). Πειραματικά & κλινικά δεδομένα δικαιολογούν επιλεκτική, προσεκτική χρήση σε παιδιά, όταν δεν υπάρχει εναλλακτική ασφαλής θεραπεία. Consensus reports of International Society of Chemotherapy. Pediatr Infect Dis J 1995;14:1-9
 Ηλεκτρολυτικές διαταραχές (υποκαλιαιμία) Καρδιολογικά προβλήματα (μοξιφλοξασίνη) Παράταση QTc, καρδιακή ανεπάρκεια με μειωμένο κλάσμα εξώθησης, κλινικά σημαντική βραδυκαρδία, ιστορικό αρρυθμίας. Ηλικία < 18 ετών, κύηση και γαλουχία. lthough fluoroquinolones are highly effective and safe therapies for adults, they have not been widely prescribed for pediatric patients principally because of laboratory studies demonstrating arthropathy in immature animals following exposure to fluoroquinolones (3, 6, 7, 27). A rather large body of information regarding fluoroquinolone use in children and adolescents for a variety of conditions has accumulated, mainly due to compassionate use in certain pediatric patient populations (1, 3, 14, 27, 29). There has been no report in the scientific literature of a definitive fluoroquinolone-associated case of arthropathy in a child despite several radiological and magnetic resonance imaging investigations (3, 8, 14, 22). Retrospective reviews and prospective studies support the view that fluoroquinolone-associated toxicity unique to children may not occur or occurs at frequencies that are extremely low (1, 3, 14, 23, 27,29). Πειραματικά & κλινικά δεδομένα δικαιολογούν επιλεκτική, προσεκτική χρήση σε παιδιά, όταν δεν υπάρχει εναλλακτική ασφαλής θεραπεία. Consensus reports of International Society of Chemotherapy. Pediatr Infect Dis J 1995;14:1-9.")
196
Κύριες κλινικές ενδείξεις κινολονών
Λοιμώξεις ουροποιητικού - προστατίτις Γαστρεντερικές λοιμώξεις Λοιμώξεις αναπνευστικού Λοιμώξεις δέρματος, μαλακών μορίων, οστών, αρθρώσεων Ενδοκοιλιακές λοιμώξεις (με μετρονιδαζόλη) Βακτηριακή μηνιγγίτιδα (μοξιφλοξασίνη, β εκλογή) Φυματίωση (δεύτερης γραμμής)
 Βακτηριακή μηνιγγίτιδα (μοξιφλοξασίνη, β εκλογή) Φυματίωση (δεύτερης γραμμής)")
197
(17th Century, National Gallery, London)
197

198
Αμινογλυκοσίδες Παραγόμενες από Micromonospora spp
Παραγόμενες από στρεπτομύκητες (Streptomycees spp) Στρεπτομυκίνη , Νεομυκίνη , Παρομομυκίνη, Καναμυκίνη Τομπραμυκίνη, Σπεκτινομυκίνη Παραγόμενες από Micromonospora spp Γενταμικίνη, Σισομικίνη Ημισυνθετικές αμινογλυκοσίδες Διβεκασίνη, Αμικασίνη, Νετιλμικίνη
 Στρεπτομυκίνη , Νεομυκίνη , Παρομομυκίνη, Καναμυκίνη. Τομπραμυκίνη, Σπεκτινομυκίνη. Παραγόμενες από Micromonospora spp. Γενταμικίνη, Σισομικίνη. Ημισυνθετικές αμινογλυκοσίδες. Διβεκασίνη, Αμικασίνη, Νετιλμικίνη.")
199
Αμινογλυκοσίδη Έτος ανακάλυψης Σκεύασμα
Αμινογλυκοσίδη Έτος ανακάλυψης Σκεύασμα Στρεπτομυκίνη Streptomycin sulfate® Νεομυκίνη Nivemycin® Παρομομυκίνη Καναμυκίνη Ftalin® Τομπραμυκίνη Nebsin® Σπεκτινομυκίνη Trobicin® Γενταμικίνη Garamycin® Σισομικίνη Geonyn® Διβεκασίνη Rolimycin® Αμικασίνη Briklin®, Νετιλμικίνη Netromycin®

200
Αντιμικροβιακό φάσμα αμινογλυκοσιδών
Gram-θετικά αερόβια S. aureus & coagulase-negative staph viridans streptococci Enterococcus sp. Gram-αρνητικά αερόβια (όχι streptomycin) E. coli, K. pneumoniae, Proteus sp., Acinetobacter, Citrobacter, Enterobacter sp. Morganella, Providencia, Serratia, Salmonella, Shigella Pseudomonas aeruginosa (amik>tobra>gent) Brucella spp, Yersinia pestis, Franciscella tularensis Μυκοβακτηρίδια (streptomycin ή amikacin) Φυματιώδη ΜΒ Μη φυματιώδη ΜΒ (άτυπα)
 E. coli, K. pneumoniae, Proteus sp., Acinetobacter, Citrobacter, Enterobacter sp. Morganella, Providencia, Serratia, Salmonella, Shigella. Pseudomonas aeruginosa (amik>tobra>gent) Brucella spp, Yersinia pestis, Franciscella tularensis. Μυκοβακτηρίδια (streptomycin ή amikacin) Φυματιώδη ΜΒ. Μη φυματιώδη ΜΒ (άτυπα)")
201
Φαρμακοκινητική αμινογλυκοσιδών
Κατανομή: Πολύ καλή σε νεφρούς & ούρα Καλή σε ασκιτικό, αρθρικό, πλευριτικό υγρό Μη ικανοποιητική σε ΕΝΥ, βρ. εκκρίσεις, οστά, χοληφόρα, προστάτη, οφθαλμό, ενδοκυτταρίως Αποβολή: σπειραματική διήθηση (επίπεδα ούρων x πλάσματος). Ημιπερίοδος ζωής: h (35-50 h σε ανουρία) σε νεφρικό φλοιό 100 h σε περι- & ενδολέμφο έσω ωτός 12 h Τ/ Σύνδεση με λεύκωμα Νεφρική κάθαρση (επίπεδα ούρων x πλάσματος) Σπειρ. διήθηση επαναρ. σε νεφρικά σωληνάρια άθροιση σε νεφρικό φλοιό T/2 σε πλάσμα: ~2 h (35-50 h επί ανουρίας) σε νεφρικό φλοιό 100 h σε περι- & ενδολέμφο έσω ωτός 12 h Distribution: Freely into the vascular space Interstitial spaces of most tissues Volume of distribution increases in edematous states and decreases in obese patients (on L/kg basis) Decreased concentrations: Bronchial secretions, CSF, biliary tract, synovial fluid, and in the eye Excreted by the kidneys Half-life: 1.5 to 3.5 hours
. Ημιπερίοδος ζωής: h (35-50 h σε ανουρία) σε νεφρικό φλοιό 100 h. σε περι- & ενδολέμφο έσω ωτός 12 h. Τ/2 Σύνδεση με λεύκωμα. Νεφρική κάθαρση (επίπεδα ούρων x πλάσματος) Σπειρ. διήθηση επαναρ. σε νεφρικά σωληνάρια άθροιση σε νεφρικό φλοιό. T/2 σε πλάσμα: ~2 h (35-50 h επί ανουρίας) σε νεφρικό φλοιό 100 h. σε περι- & ενδολέμφο έσω ωτός 12 h. Distribution: Freely into the vascular space. Interstitial spaces of most tissues. Volume of distribution increases in edematous states and decreases in obese patients (on L/kg basis) Decreased concentrations: Bronchial secretions, CSF, biliary tract, synovial fluid, and in the eye. Excreted by the kidneys. Half-life: 1.5 to 3.5 hours.")
202
Φαρμακοδυναμική αμινογλυκοσιδών
Ταχεία μικροβιοκτόνος δράση εξαρτώμενη από συγκέντρωση και pH (MIC πενταπλάσια σε pH <6,5) Η κλινική ανταπόκριση είναι ανάλογη του πηλίκου Cmax:MIC (π.χ. Cmax:MIC 883% 10 80% 12: 92%) Μεταντιβιοτική δραστικότητα 3 ώρες (1-7,5)
 Η κλινική ανταπόκριση είναι ανάλογη του πηλίκου Cmax:MIC (π.χ. Cmax:MIC 883% 10 80% 12: 92%) Μεταντιβιοτική δραστικότητα 3 ώρες (1-7,5)")
203
Ανεπιθύμητες ενέργειες αμινογλυκοσιδών
Νεφροτοξικότητα (5-10%) Ωτοτοξικότητα (5-7%) Αλλεργικές αντιδράσεις (1-2%) Αιματολογικές διαταραχές (σπάνια) Nευρομυικός αποκλεισμός (πολύ σπάνια) Άλλες σπάνιες.
 Ωτοτοξικότητα (5-7%) Αλλεργικές αντιδράσεις (1-2%) Αιματολογικές διαταραχές (σπάνια) Nευρομυικός αποκλεισμός (πολύ σπάνια) Άλλες σπάνιες.")
204
Κλινικές ενδείξεις αμινογλυκοσιδών
Κλινικές ενδείξεις αμινογλυκοσιδών Λοιμώξεις ουροποιητικού Βακτηριακή ενδοκαρδίτιδα Σήψη Ενδοκοιλιακές λοιμώξεις Νοσοκομειακή πνευμονία Εμπύρετος ουδετεροπενία Λοιμώξεις από λιστέρια + β λακταμικό

205
Δοσολογία αμινογλυκοσιδών
Γενταμικίνη Τομπραμυκίνη Νετιλμικίνη Αμικασίνη Στρεπτομυκ. 3-5,5 (7) mg/kg/24h 4-6 (7,5) mg/kg/24h DOSING Current practice is to administer the total daily dose as a single injection, which is associated with less toxicity and equal efficacy as multiple-dose regimens. This diminished toxicity is probably due to a threshold effect from accumulation of drug in the inner ear or in the kidney. Despite the higher peak concentration, a once-daily dosing regimen provides a longer period when concentrations fall below the threshold for toxicity than does a multiple-dose regimen (12 hours vs. less than 3 hours total in Figure 45–3), accounting for its lower toxicity. Aminoglycoside bactericidal activity, on the other hand, is related directly to the peak concentration achieved because of concentrationdependent killing and a concentration-dependent postantibiotic effect. Once-daily regimens are safer with equal efficacy, cost less, and are administered more easily. Exceptions include use in pregnancy, neonatal and pediatric infections, and low-dose combination therapy of bacterial endocarditis. Once-daily dosing also should be avoided in patients with creatinine clearances of <20–25 mL/min, where dosing every 48 hours is more appropriate. Whether once-daily or multiple-daily dosing is used, the dose must be adjusted for patients with creatinine clearances of <80–100 mL/min, and plasma concentrations must be monitored. Nomograms may be helpful in selecting initial doses, but variability in aminoglycoside clearance among patients is too large for these to be relied on for more than a few days. If a patient likely will be treated with an aminoglycoside for more than 3–4 days, then plasma concentrations should be monitored. For twice- or thrice-daily dosing regimens, both peak (30 minutes after dosing) and trough (immediately before the next dose) plasma concentrations are determined. The peak concentration documents that the dose produces therapeutic concentrations, while the trough concentration is used to avoid toxicity. With once-daily regimens, the trough concentration is either measured directly or estimated using various nomograms; a trough concentration >2 mg/mL predicts toxicity. 15 mg/kg/24h
 mg/kg/24h. 4-6 (7,5) mg/kg/24h. DOSING. Current practice is to administer the total daily dose as a single injection, which is associated with less toxicity and equal efficacy as multiple-dose regimens. This diminished toxicity is probably due to a threshold effect from accumulation of drug in the inner ear or in the kidney. Despite the higher. peak concentration, a once-daily dosing regimen provides a longer period when concentrations fall below the threshold for toxicity than does a multiple-dose regimen (12 hours vs. less than 3 hours total in Figure 45–3), accounting for its lower toxicity. Aminoglycoside bactericidal activity, on the other hand, is related directly to the peak concentration achieved because of concentrationdependent. killing and a concentration-dependent postantibiotic effect. Once-daily regimens are safer with equal efficacy, cost less, and are administered more easily. Exceptions include use in pregnancy, neonatal and pediatric infections, and low-dose combination therapy of bacterial endocarditis. Once-daily dosing also should be avoided in patients with creatinine clearances of <20–25 mL/min, where dosing every 48 hours is more appropriate. Whether once-daily or multiple-daily dosing is used, the dose must be adjusted for patients with. creatinine clearances of <80–100 mL/min, and plasma concentrations must be monitored. Nomograms. may be helpful in selecting initial doses, but variability in aminoglycoside clearance among. patients is too large for these to be relied on for more than a few days. If a patient likely will be treated. with an aminoglycoside for more than 3–4 days, then plasma concentrations should be monitored. For twice- or thrice-daily dosing regimens, both peak (30 minutes after dosing) and trough. (immediately before the next dose) plasma concentrations are determined. The peak concentration. documents that the dose produces therapeutic concentrations, while the trough concentration is. used to avoid toxicity. With once-daily regimens, the trough concentration is either measured. directly or estimated using various nomograms; a trough concentration >2 mg/mL predicts toxicity. 15 mg/kg/24h.")
206
Goodman and Gilman’s Clinical Pharmacology Copyright © 2008 by The McGraw-Hill Companies, Inc

207
Κανόνες δοσολογίας αμινογλυκοσιδών
Χορήγηση με βάση το ΣΒ Σε παχύσαρκους υπολογίζεται το 50% του επιπλέον βάρους Τακτικός προσδιορισμός επιπέδων ορού Προσαρμογή δοσολογίας σε ΝΑ Χορήγηση άπαξ ημερησίως Δεν υπάρχουν επαρκή στοιχεία ή πρέπει να αποφεύγεται η άπαξ ημερησίως για Λοιμώδη ενδοκαρδίτιδα Εμπύρετη ουδετεροπενία (στηρίζεται από δεδομένα σε μη ουδετεροπενικούς) Κυστική ίνωση Κύηση Παιδιά και νεογνά
 Κυστική ίνωση. Κύηση. Παιδιά και νεογνά.")
208
-εφάπαξ ημερησίως (7,0 mg/kg) (κενοί κύκλοι) ή
Αποτελεσματικότητα γενταμυκίνης έναντι παθογόνου με MIC 2 mg/L σε δοσολογικό σχήμα -εφάπαξ ημερησίως (7,0 mg/kg) (κενοί κύκλοι) ή -1,5 mg/kg/8h (πλήρεις κύκλοι) Απαιτούμενο IQ= Cmax/MIC=10-12
 (κενοί κύκλοι) ή. -1,5 mg/kg/8h (πλήρεις κύκλοι) Απαιτούμενο IQ= Cmax/MIC=")
209
Επίπεδα πλάσματος γενταμυκίνης μετά από χορήγηση 5,1 mg/kg ημερησίως
Η νεφρο- και ωτο-τοξικότητα είναι χρονοεξερτώμενη και όχι δοσοεξαρτώμενη

210
Θεραπευτικά επίπεδα αμινογλυκοσιδών (μg/L)
Επίπεδα Κορυφής Επίπεδα Κοιλάδας Σοβαρές λοιμώξεις Επικίνδυνες λοιμώξεις Γενταμ. 6-8 8-10 0.5- 1 1-2 Τομπρ. Νετιλμ. 8-10 (20-30) Aμικασ. 20-25 25-30 1-4 4-8 Χορήγηση 3 φορές/d για γενταμικίνη, τομπραμυκίνη, νετιλμικίνη και 2 φορές/d για αμικασίνη (σε παρένθεση για εφάπαξ ημερήσια χορήγηση) In patients with normal renal function, defined peak (5-10 mg/L) and trough levels (less than 2 mg/L) for gentamicin, tobramicin, and netilmicin are considered therapeutic. Netilmicin peak and trough levels were investigated in 50 patients requiring hemodialysis due to acute (70%) or permanent (30%) renal failure. Netilmicin was given at a dosage interval of 24 h, with a loading dose on the first day (1.5 mg/kg) and a reduced daily maintenance dose (0.5 mg/kg) supplemented to the posthemodialysis dosage (1.3 mg/kg) after each hemodialysis. As compared with studies on patients not requiring hemodialysis, mortality (44%) was higher, mainly due to uncontrolled infection, whereas ototoxicity (17%) was not. Peak (5.9 +/- 1.7 mg/L) and trough plasma levels (3.0 +/- 0.9 mg/L) were significantly lower in patients who did not respond and died than were peak (8.2 +/- 2.5 mg/L) and trough (3.8 +/- 1.2 mg/L) levels in patients responding to aminoglycoside treatment. In renal failure patients, there is obviously not only the risk of overdosing and toxic side effects but also the risk of insufficient bactericidal effect as a result of underdosing. Consequently, by use of an aminoglycoside aminoglycoside dosage similar to the present schedule, peak levels (5-10 mg/L) as desired in normal subjects but trough levels (2.5-5 mg/L) that are considerably higher than in normal subjects should be the target concentrations for patients with advanced renal failure.
 Aμικασ Χορήγηση 3 φορές/d για γενταμικίνη, τομπραμυκίνη, νετιλμικίνη και 2 φορές/d για αμικασίνη (σε παρένθεση για εφάπαξ ημερήσια χορήγηση) In patients with normal renal function, defined peak (5-10 mg/L) and trough levels (less than 2 mg/L) for gentamicin, tobramicin, and netilmicin are considered therapeutic. Netilmicin peak and trough levels were investigated in 50 patients requiring hemodialysis due to acute (70%) or permanent (30%) renal failure. Netilmicin was given at a dosage interval of 24 h, with a loading dose on the first day (1.5 mg/kg) and a reduced daily maintenance dose (0.5 mg/kg) supplemented to the posthemodialysis dosage (1.3 mg/kg) after each hemodialysis. As compared with studies on patients not requiring hemodialysis, mortality (44%) was higher, mainly due to uncontrolled infection, whereas ototoxicity (17%) was not. Peak (5.9 +/- 1.7 mg/L) and trough plasma levels (3.0 +/- 0.9 mg/L) were significantly lower in patients who did not respond and died than were peak (8.2 +/- 2.5 mg/L) and trough (3.8 +/- 1.2 mg/L) levels in patients responding to aminoglycoside treatment. In renal failure patients, there is obviously not only the risk of overdosing and toxic side effects but also the risk of insufficient bactericidal effect as a result of underdosing. Consequently, by use of an aminoglycoside aminoglycoside dosage similar to the present schedule, peak levels (5-10 mg/L) as desired in normal subjects but trough levels (2.5-5 mg/L) that are considerably higher than in normal subjects should be the target concentrations for patients with advanced renal failure.")
211
MΑΚΡΟΛΙΔΙΚΑ ΑΝΤΙΒΙΟΤΙΚΑ
Παλαιότερα Picromycin (1950) Erythromycin (1952) Spiramycin Carbomycin Kitasamycin Oleandomycin Josamycin Νεότερα Clarithromycin Azithromycin Roxithromycin Dirithromycin Flurithromycin Darithromycin
 Erythromycin (1952) Spiramycin. Carbomycin. Kitasamycin. Oleandomycin. Josamycin. Νεότερα. Clarithromycin. Azithromycin. Roxithromycin. Dirithromycin. Flurithromycin. Darithromycin.")
212
Αντιμικροβιακό φάσμα μακρολιδίων
Gram-θετικά (Clarithro>Erythro>Azithro) S. pyogenes, S. viridans, S. pneumoniae (μόνο PSSP, συχνή αντοχή) S. aureus (MSSA) Corynebacterium sp. Gram-αρνητικά (Azithro>Clarithro>Erythro) Neiseria spp (συχνή αντοχή), H. influenzae (μέτρια δραστικότητα, όχι Ερυθρο.) M. catarrhalis Άτυπα (όλα άριστη δραστικότητα) Chlamydia spp. Mycoplasma spp. Legionella pneumophila, Ureaplasma urealyticum Άλλα βακτήρια Mycobacterium avium complex, Mycobacterium leprae (όχι Ερυθρο.) Treponema pallidum, Borrelia, Campylobacter, Helicobacter pylori, Bordetella pertussis, Pasteurella Αναερόβια στόματος
 S. pyogenes, S. viridans, S. pneumoniae (μόνο PSSP, συχνή αντοχή) S. aureus (MSSA) Corynebacterium sp. Gram-αρνητικά. (Azithro>Clarithro>Erythro) Neiseria spp (συχνή αντοχή), H. influenzae (μέτρια δραστικότητα, όχι Ερυθρο.) M. catarrhalis. Άτυπα. (όλα άριστη δραστικότητα) Chlamydia spp. Mycoplasma spp. Legionella pneumophila, Ureaplasma urealyticum. Άλλα βακτήρια. Mycobacterium avium complex, Mycobacterium leprae. (όχι Ερυθρο.) Treponema pallidum, Borrelia, Campylobacter, Helicobacter pylori, Bordetella pertussis, Pasteurella. Αναερόβια στόματος.")
213
Φαρμακοκινητική Μακρολιδίων
Ερυθρομυκίνη Κλαριθρομυκίνη Αζιθρομυκίνη Απορόφηση 15-45% (εστέρες και άλατα εστέρων) 55% οξεάντοχος, ανεξαρτήτως τροφής 38% οξεάντοχος, απορ- ρόφηση caps με τροφή. Κατανομή Πολύ καλή διείσδυση σε ιστούς (αναπνευστικό, προστάτης) και κύτταρα (ΠΜΠ), κυρίως για clari. & azithro Ελάχιστη διείσδυσε σε ΕΝΥ Αποβολή Ηπατική Νεφρική 5% Ηπατική (CYP 3A4 14-OH-clarithromycin) Νεφρική 30-40% (1/2 δόσης εάν CrCl < 30) Χολή (μικρός ηπατικός μεταβ) Νεφρική 12% Ημιπ. ζωής 1,6 (8 h σε ηπατ ανεπ.) 5-7 h 2-72 h Σύνδ. με λεύκωμα 70% 60-70% 7-50% Επίπεδα στο αίμα χαμηλά (Per Os: 0,4 μg/ml , ΕΦ: 3,63+1,6 μg/ml) Επίπεδα σε ιστούς: 50x υψηλότερα είσοδος σε κύτταρα t/2 σε ιστούς 2-4 ημέρες. Κλαριθρ 14-μελής λακτονικός δακτύλιος methoxyl group στη θέση C-6 Σταθερότητα σε όξινο περιβάλλον Μερικός μεταβολισμός από σε 14-OH-clarithromycin Αποβολή από νεφρούς 30-40% (Εάν CrCl < 30 ml/min: ½ δόσης) Ημιπερίοδος ζωής 5-7 ώρες Βιοδιαθεσιμότητα: 55% Δέσμευση με λευκώματα: 70% Hμιπερίοδος ζωής:
 55% οξεάντοχος, ανεξαρτήτως τροφής. 38% οξεάντοχος, απορ- ρόφηση caps με τροφή. Κατανομή. Πολύ καλή διείσδυση σε ιστούς (αναπνευστικό, προστάτης) και κύτταρα (ΠΜΠ), κυρίως για clari. & azithro. Ελάχιστη διείσδυσε σε ΕΝΥ. Αποβολή. Ηπατική. Νεφρική 5% Ηπατική (CYP 3A4 14-OH-clarithromycin) Νεφρική 30-40% (1/2 δόσης εάν CrCl < 30) Χολή (μικρός ηπατικός μεταβ) Νεφρική 12% Ημιπ. ζωής. 1,6 (8 h σε ηπατ ανεπ.) 5-7 h h. Σύνδ. με λεύκωμα. 70% 60-70% 7-50% Επίπεδα στο αίμα χαμηλά (Per Os: 0,4 μg/ml , ΕΦ: 3,63+1,6 μg/ml) Επίπεδα σε ιστούς: 50x υψηλότερα. είσοδος σε κύτταρα t/2 σε ιστούς 2-4 ημέρες. Κλαριθρ. 14-μελής λακτονικός δακτύλιος. methoxyl group στη θέση C-6. Σταθερότητα σε όξινο περιβάλλον. Μερικός μεταβολισμός από σε 14-OH-clarithromycin. Αποβολή από νεφρούς 30-40% (Εάν CrCl < 30 ml/min: ½ δόσης) Ημιπερίοδος ζωής 5-7 ώρες. Βιοδιαθεσιμότητα: 55% Δέσμευση με λευκώματα: 70% Hμιπερίοδος ζωής:")
214
Ανεπιθύμητες ενέργειες μακρολιδίων
Από ΓΕΣ: έως 33 % Ναυτία, έμετοι, διάρροια, δυσπεψία Συχνότερα με erythro, σπανιότερα με νεότερα Χολοστατική ηπατίτιδα: σπάνια 1 - 2 εβδ. μετά από θεραπεία με erythromycin estolate Θρομβοφλεβίτιδα: συχνή σε IV Erythro> Clari.>Azithro Αραίωση, βραδεία έγχυση Άλλες: Ωτοτοξικότητα (υψηλή δόση erythro) Παράταση QTc Αλλεργία
 Παράταση QTc. Αλλεργία.")
215
Φαρμακευτικές αλληλεπιδράσεις μακρολιδίων
Με στατίνες κίνδυνος ραβδομυόλυσης Με κορτικοστεροειδή μείωση επιπέδων κορτικοστεροειδών Με φάρμακα που παρατείνουν το QT κίνδυνος αρρυθμίας Erythromycin & Clarithromycin αναστολή CY p450 επιπέδων: Theophylline, Digoxin, Disopyramide, Carbamazepine, Valproic acid, Phenytoin, Cyclosporine, Terfenadine, Astemizole, Cisapride, Warfarin, Ergot alkaloids ΑRT (με NNRTI , ενώ με PI επιπ. κλαριθρομυκίνης) Azithromycin: κυρίως με cyclosporine & tacrolimus Με θεοφυλλίνη, καφεΐνη, κουμαρινικά, διγοξίνη, κυκλοσπορίνη, καρβαμαζεπίνη Με στατίνες κίνδυνος ραβδομυόλυσης Με κορτικοστεροειδή ( επιπέδων) Με φάρμακα που παρατείνουν το QT Με χλωραμφενικόλη ή λινκομυκίνη ( δραστικότητας).
 Azithromycin: κυρίως με cyclosporine & tacrolimus. Με θεοφυλλίνη, καφεΐνη, κουμαρινικά, διγοξίνη, κυκλοσπορίνη, καρβαμαζεπίνη Με στατίνες κίνδυνος ραβδομυόλυσης. Με κορτικοστεροειδή ( επιπέδων) Με φάρμακα που παρατείνουν το QT. Με χλωραμφενικόλη ή λινκομυκίνη ( δραστικότητας).")
216
ΓΕΝΙΚΑ ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΜΑΚΡΟΛΙΔΙΩΝ
Άριστες ιστικές συγκεντρώσεις (>πλάσματος) Διείσδυση: αναπνευστικό, αμυγδαλές προστάτης ΠΜΠ λευκοκύτταρα (σημαντικό για Chlamydia και Legionella sp) Τύπος αντιμικροβιακής δράσης: time dependent Η δραστικότητα καθορίζεται από χρόνο παραμονής συγκέντρωσης αντιβιοτικού υψηλότερα MIC
 Διείσδυση: αναπνευστικό, αμυγδαλές. προστάτης. ΠΜΠ λευκοκύτταρα (σημαντικό για Chlamydia και Legionella sp) Τύπος αντιμικροβιακής δράσης: time dependent. Η δραστικότητα καθορίζεται από χρόνο παραμονής συγκέντρωσης αντιβιοτικού υψηλότερα MIC.")
217
Κύριες κλινικές ενδείξεις μακρολιδίων
Λοίμωξη αν. αναπνευστικού, πνευμονία κοινότητας Κυρίως από Legionella (αζιθρ), Chlamydia, Mycoplasma spp Μη ειδική ουρηθρίτιδα Μη επιπλεγμένες λοιμώξεις δέρματος Εντερική λοίμωξη από Campylobacter Λοίμωξη από μη φυματιώδη ΜΒ (M. avium) Κοκύτης, διφθερίτιδα (ερυθρ) Λοίμωξη από Η. Pylori Στρεπτοκοκκική φαρυγοαμυγδαλίτιδα (αλλεργία σε πενικιλλίνη) Τέτανος (αλλεργία σε πενικιλλίνη)
, Chlamydia, Mycoplasma spp. Μη ειδική ουρηθρίτιδα. Μη επιπλεγμένες λοιμώξεις δέρματος. Εντερική λοίμωξη από Campylobacter. Λοίμωξη από μη φυματιώδη ΜΒ (M. avium) Κοκύτης, διφθερίτιδα (ερυθρ) Λοίμωξη από Η. Pylori. Στρεπτοκοκκική φαρυγοαμυγδαλίτιδα (αλλεργία σε πενικιλλίνη) Τέτανος (αλλεργία σε πενικιλλίνη)")
218
Διαφορές μακρολιδίων Erythromycin Clarithromycin Azithromycin
Συχνές διαταραχές από ΓΕΣ, πολλές αλληλεπιδράσεις Πολύ χαμηλότερο κόστος Clarithromycin Δεν προκαλεί κοιλιακό άλγος Επιτυγχάνει υψηλότερες συγκεντρώσεις στο αίμα (500 mg per Os: 2,5-5 μg/ml) Χαμηλότερη δοσολογία, σε αραιότερα διαστήματα Δραστικότερη ερυθρομυκίνης έναντι gram αρν και άτυπων Αλληλεπιδράσεις Αθροίζεται σε νεφρική ανεπάρκεια Azithromycin Λιγότερο δραστική ερυθρο. και κλαριθρο. έναντι Gram-θετ. Δραστικότερη έναντι Gram-αρν. και άτυπων Χαμηλή συγκέντρωση στο αίμα (Per Os: 0,4-0,8 μg/ml, ΕΦ: 3,63+1,6 μg/ml) Υψηλή συγκέντρωση σε ιστούς (έως 50x, t/2 2-4 ημέρες)
 Χαμηλότερη δοσολογία, σε αραιότερα διαστήματα. Δραστικότερη ερυθρομυκίνης έναντι gram αρν και άτυπων. Αλληλεπιδράσεις. Αθροίζεται σε νεφρική ανεπάρκεια. Azithromycin. Λιγότερο δραστική ερυθρο. και κλαριθρο. έναντι Gram-θετ. Δραστικότερη έναντι Gram-αρν. και άτυπων. Χαμηλή συγκέντρωση στο αίμα (Per Os: 0,4-0,8 μg/ml, ΕΦ: 3,63+1,6 μg/ml) Υψηλή συγκέντρωση σε ιστούς (έως 50x, t/2 2-4 ημέρες)")
220
Κετολίδη (συγγενής προς μακρολίδες) Αντιμικροβιακό φάσμα
Telithromycin Κετολίδη (συγγενής προς μακρολίδες) Αντιμικροβιακό φάσμα Group A, B, C, G Streptococci, S. pneumoniae, Σταφυλόκοκκοι (ανθεκτικά στελέχη) Chlamydia spp., Mycoplasma spp., Legionella pneumophila M. catarrhalis, H. influenzae, Bordetella pertussis Ενδείξεις Πνευμονία κοινότητας (κίνδυνος ηπατοτοξικότητας) Telithromycin is approved for use against bacterial respiratory infections Active against most strains of Streptococcus pneumoniae, including penicillin- and macrolide-resistant strains Also active against more strains of Staphylococci Only available in oral formulation
 Αντιμικροβιακό φάσμα. Group A, B, C, G Streptococci, S. pneumoniae, Σταφυλόκοκκοι (ανθεκτικά στελέχη) Chlamydia spp., Mycoplasma spp., Legionella pneumophila. M. catarrhalis, H. influenzae, Bordetella pertussis. Ενδείξεις. Πνευμονία κοινότητας (κίνδυνος ηπατοτοξικότητας) Telithromycin is approved for use against bacterial respiratory infections. Active against most strains of Streptococcus pneumoniae, including penicillin- and macrolide-resistant strains. Also active against more strains of Staphylococci. Only available in oral formulation.")
221
Φυσικά παράγωγα στρεπτομυκήτων
TETΡΑΚΥΚΛΙΝΕΣ Φυσικά παράγωγα στρεπτομυκήτων Οξυτετρακυκλίνη, τετρακυκλίνη, διμεκλοτετρακυκλίνη Παραγόμενες ημισυνθετικώς Δοξυκυκλίνη, μινοκυκλίνη.

222
ΦΑΡΜΑΚΟΚΙΝΗΤΙΚΕΣ ΙΔΙΟΤΗΤΕΣ
Aπορρόφηση από ΓΕΣ oxytetracycline, tetracycline: ατελής ( με γαλακτοκομικά, άλατα Al, Ca, Mg, Fe, Zn, Bi) doxycycline, minocycline: πλήρης Ημιπερίοδος ζωής 6–12 h για oxytetracycline, tetracycline 16-18 h για doxycycline, minocycline t/2 doxycycline από βαρβιτουρικά, φενυντοϊνη, ριφαμπικίνη (CYP) Κατανομή Ούρα, προστάτης, ΕΝΥ, ΔΕΣ, οστά, πλακούντας, αμνιακό υγρό, γάλα κλπ Απέκκριση Νεφρική (doxycycline δια κοπράνων δεν αθροίζεται σε ΝΑ) Αποβολή με χολή, αλλά επαναρρόφηση
 doxycycline, minocycline: πλήρης. Ημιπερίοδος ζωής. 6–12 h για oxytetracycline, tetracycline h για doxycycline, minocycline. t/2 doxycycline από βαρβιτουρικά, φενυντοϊνη, ριφαμπικίνη (CYP) Κατανομή. Ούρα, προστάτης, ΕΝΥ, ΔΕΣ, οστά, πλακούντας, αμνιακό υγρό, γάλα κλπ. Απέκκριση. Νεφρική (doxycycline δια κοπράνων δεν αθροίζεται σε ΝΑ) Αποβολή με χολή, αλλά επαναρρόφηση.")
223
Αντιμικροβιακό φάσμα τετρακυκλινών
Gram θετικοί και αρνητικοί κόκκοι Στρεπτόκοκκοι, Σταφυλόκοκκοι, Ναϊσέριες Gram θετικοί και αρνητικοί βάκιλλοι Bacillus anthracis, Brucella spp., Haemophilus spp. Campylobacter jejuni, Vibrio cholera, Yersinia spp Escherichia coli, Proteus spp, Serratia spp. Pseudomonas pseudomallei, Francisella tularensis Bordetella pertusis, Branchamela catarrhalis, Listeria Αναερόβιοι Gram αρνητικοί βάκιλλοι: Fusobacterium, Bacteroides spp (50%). Μυκοπλάσματα, ρικέτσιες, χλαμύδια, Legionella spp Σπειροχαίτες (T. pallidum, B. burgdorferi) Άτυπα μυκοβακτηρίδα (ορισμένα) Αμοιβάδα, Πλασμώδια ελονοσίας.
. Μυκοπλάσματα, ρικέτσιες, χλαμύδια, Legionella spp. Σπειροχαίτες (T. pallidum, B. burgdorferi) Άτυπα μυκοβακτηρίδα (ορισμένα) Αμοιβάδα, Πλασμώδια ελονοσίας.")
224
Κλινικές ενδείξεις τετρακυκλινών
Λοιμώξεις από Rickettsia, Mycoplasma, Chlamydia spp Βρουκέλλωση (με στρεπτομυκίνη ή ριφαμπικίνη) Λοιμώξεις από σπειροχαίτες (Lyme, σύφιλη σε αδυναμία χορήγησης πενικιλίνης ) Μη γονοκοκκική ουρηθρίτιδα Λοιμώξεις αναπνευστικού Άνθραξ (δερματικός, πρόληψη ή θεραπεία) Χολέρα (300 mg doxycycline εφάπαξ) Διάρροια ταξιδιωτών (συχνά ανθεκτικά παθογόνα) Λεπτοσπείρωση Τουλαραιμία, Ακτινομύκωση Ακμή
 Λοιμώξεις από σπειροχαίτες (Lyme, σύφιλη σε αδυναμία χορήγησης πενικιλίνης ) Μη γονοκοκκική ουρηθρίτιδα. Λοιμώξεις αναπνευστικού. Άνθραξ (δερματικός, πρόληψη ή θεραπεία) Χολέρα (300 mg doxycycline εφάπαξ) Διάρροια ταξιδιωτών (συχνά ανθεκτικά παθογόνα) Λεπτοσπείρωση. Τουλαραιμία, Ακτινομύκωση. Ακμή.")
225
Ανεπιθύμητες ενέργειες τετρακυκλινών
Φωτοδερματίτιδα Αλλοίωση εντερικής χλωρίδας, μυκητιάσεις, υπερλοιμώξεις Γαστρεντερικές διαταραχές, ψευδομεμβρανώδης κολίτιδα Ηπατοτοξικότητα (υψηλές δόσεις, εγκυμοσύνη) Χρώση δοντιών (καφέ, μόνιμη) σε παιδιά (κυρίως 2 μηνών -5 ετών) Αναστολή ανάπτυξη σκελετού σε πρόωρα Αιματολογικές διαταραχές (σε μακρά χορήγηση) Αλλεργικές αντιδράσεις (σπάνιες) Βλάβη αιθoυσαίου ν. (μινοκυκλίνη, 35-70%). Νεφροτοξικότητα (σπάνια, αζωθαιμία λόγω καταβολισμού)
 Χρώση δοντιών (καφέ, μόνιμη) σε παιδιά (κυρίως 2 μηνών -5 ετών) Αναστολή ανάπτυξη σκελετού σε πρόωρα. Αιματολογικές διαταραχές (σε μακρά χορήγηση) Αλλεργικές αντιδράσεις (σπάνιες) Βλάβη αιθoυσαίου ν. (μινοκυκλίνη, 35-70%). Νεφροτοξικότητα (σπάνια, αζωθαιμία λόγω καταβολισμού)")
227
ΤΡΟΠΟΙ ΧΟΡΗΓΗΣΗΣ - ΔΟΣΟΛΟΓΙΑ
Μπορούν να χορηγηθούν per Os, παρεντερικώς, τοπικώς Δόση (per Os) Τετρακυκλίνες: g/day σε ενήλικες Δοξυκυκλίνη: 100 mg /12 h (με νερό, όρθια θέση για 30 min) Μινοκυκλίνη: 200 mg/d αρχικά, έπειτα 100 mg /12 h
 Τετρακυκλίνες: g/day σε ενήλικες. Δοξυκυκλίνη: 100 mg /12 h (με νερό, όρθια θέση για 30 min) Μινοκυκλίνη: 200 mg/d αρχικά, έπειτα 100 mg /12 h.")
228
Tigecycline (Glycylcycline, από μινοκυκλίνη)
Ευρύ φάσμα Gram-θετικά (+ ΜRSA, VISA, όχι VRE) Gram-αρνητικά Haemophilus influenzae, Neisseria spp. Enterobacteriaceae ΔΕΝ είναι δραστική έναντι Pseudomonas και Proteus spp Αναερόβια (+Bacteroides fragilis) Άτυπα: Mycoplasma spp. (Legionella;) Ενδείξεις Επιπλεγμένες λοιμώξεις δέρματος και μαλακών μορίων Ενδοκοιλιακές λοιμώξεις Πνευμονία κοινότητας από πνευμονιόκοκκο (FDA, 2009)
 Gram-αρνητικά. Haemophilus influenzae, Neisseria spp. Enterobacteriaceae. ΔΕΝ είναι δραστική έναντι Pseudomonas και Proteus spp. Αναερόβια (+Bacteroides fragilis) Άτυπα: Mycoplasma spp. (Legionella;) Ενδείξεις. Επιπλεγμένες λοιμώξεις δέρματος και μαλακών μορίων. Ενδοκοιλιακές λοιμώξεις. Πνευμονία κοινότητας από πνευμονιόκοκκο (FDA, 2009)")
229
Ανεπιθύμητες ενέργειες Tigecycline
Ναυτία, διάρροια (έως 10%) Υπέρταση, οίδημα (2-10%) Κεφαλαλγία (6%), ζάλη (4%), αϋπνία (2%) Πυρετός, κνησμός (3%), εξάνθημα (2%) Υπολευκωματιναιμία (5%), υπεργλυκαιμία (2%), υποκαλιαιμία (2%) Θρομβοπενία (6%), αναιμία (4%), λευκοκυττάρωση (4%) Αύξηση SGPT (6%), SGOT (4%), αλκ. φωσφατάσης (4%), αμυλάσης (3%), χολερυθρίνης (2%), LDH (4%), BUN (2%) Μυική αδυναμία (3%), βήχας (4%), δύσπνοια (3%) Παγκρεατίτιδα Βαριά ηπατική βλάβη
 Υπέρταση, οίδημα (2-10%) Κεφαλαλγία (6%), ζάλη (4%), αϋπνία (2%) Πυρετός, κνησμός (3%), εξάνθημα (2%) Υπολευκωματιναιμία (5%), υπεργλυκαιμία (2%), υποκαλιαιμία (2%) Θρομβοπενία (6%), αναιμία (4%), λευκοκυττάρωση (4%) Αύξηση SGPT (6%), SGOT (4%), αλκ. φωσφατάσης (4%), αμυλάσης (3%), χολερυθρίνης (2%), LDH (4%), BUN (2%) Μυική αδυναμία (3%), βήχας (4%), δύσπνοια (3%) Παγκρεατίτιδα. Βαριά ηπατική βλάβη.")
230
Δοσολογία Tigecycline
1η δόση: 100 mg, ακολούθως 50 mg q12h iv σε έγχυση 1-3 h Δεν απαιτείται προσαρμογή δοσολογίας σε νεφρική ανεπάρκεια Σε ηπατική ανεπάρκεια Child Pugh Α & B δεν απαιτείται προσαρμογή, ενώ σε Child Pugh C μειώνεται η δοσολογία σε 25 mg q12h (αρχική δόση παραμένει 100 mg).
.")
231
ΓΛΥΚΟΠΕΠΤΙΔΙΚΑ ΑΝΤΙΒΙΟΤΙΚΑ
Βανκομυκίνη 1956, από Streptomyces orientalis Τεϊκοπλανίνη Actinoplanes teichomyceticus Mικροβιοκτόνα, δραστικά μόνο έναντι gram θετικών βακτηρίων (+MRSA) Αναστέλλουν τη σύνθεση του κυτταρικού τοιχώματος Φάρμακα εκλογής για σοβαρές MRSA λοιμώξεις
 Αναστέλλουν τη σύνθεση του κυτταρικού τοιχώματος. Φάρμακα εκλογής για σοβαρές MRSA λοιμώξεις.")
232
Φαρμακοκινητική γλυκοππετιδίων
Βανκομυκίνη Τεϊκοπλανίνη Απέκκριση Νεφρική (90%) νεφρική Σύνδεση με λευκώματα 30% 90-95% Ημιπερίοδος ζωής 6 ώρες (> 200 ώρες σε ανουρία) 7-14 ώρες (έως 150 h) Κατανομή Καρδιά, ήπαρ, νεφρούς, αποστηματικές κοιλότητες, πλευριτικό, αρθρικό, περικαρδιακό υγρό. ΕΝΥ: 10-20% (σε φλεγμονή). Νεφρική αποβολή Absorption absorption from GI tract is negligible after oral administration except in patients with intense colitis Use IV therapy for treatment of systemic infection Distribution widely distributed into body tissues and fluids, including adipose tissue; use TBW for dosing inconsistent penetration into CSF, even with inflamed meninges Elimination primarily eliminated unchanged by the kidney via glomerular filtration Απέκκριση: Ημιπερίοδος ζωής: Κατανομή: Καρδιά, ήπαρ, νεφρούς, αποστηματικές κοιλό-τητες, πλευριτικό, αρθρικό, περικαρδιακό υγρό.
 νεφρική. Σύνδεση με λευκώματα. 30% 90-95% Ημιπερίοδος ζωής. 6 ώρες. (> 200 ώρες σε ανουρία) 7-14 ώρες (έως 150 h) Κατανομή. Καρδιά, ήπαρ, νεφρούς, αποστηματικές κοιλότητες, πλευριτικό, αρθρικό, περικαρδιακό υγρό. ΕΝΥ: 10-20% (σε φλεγμονή). Νεφρική αποβολή. Absorption. absorption from GI tract is negligible after oral administration except in patients with intense colitis. Use IV therapy for treatment of systemic infection. Distribution. widely distributed into body tissues and fluids, including adipose tissue; use TBW for dosing. inconsistent penetration into CSF, even with inflamed meninges. Elimination. primarily eliminated unchanged by the kidney via glomerular filtration. Απέκκριση: Ημιπερίοδος ζωής: Κατανομή: Καρδιά, ήπαρ, νεφρούς, αποστηματικές κοιλό-τητες, πλευριτικό, αρθρικό, περικαρδιακό υγρό.")
233
Αντιμικροβιακό φάσμα γλυκοπεπτιδίων
Gram θετικοί κόκκοι Σταφυλόκοκκοι, στρεπτόκοκκοι, εντερόκοκκοι Peptococcus, Peptostreptococcus Gram θετικοί βάκιλλοι Clostridium sp. (+ C. difficile) Corynobacterium, Listeria monocytogenes, Actinomyces, Propionibacterium acnes Bacillus anthracis. Απουσία δραστικότητας έναντι gram-αρνητικών αερόβιων ή αναερόβιων
 Corynobacterium, Listeria monocytogenes, Actinomyces, Propionibacterium acnes. Bacillus anthracis. Απουσία δραστικότητας έναντι gram-αρνητικών αερόβιων ή αναερόβιων.")
234
ΑΝΤΟΧΗ ΣΤΑ ΓΛΥΚΟΠΕΠΤΙΔΙΑ
E. faecium & E. faecalis (VRE) Φαινότυποι Van A, λιγότερο Van B Μετάδοση με transposon και σε άλλα gram + Αντοχή και σε αμινογλυκοσίδες, αμπικιλλίνη S. aureus & CNS (VRSA ή VISA) Συχνά προηγείται heteroresistance Μικρός αριθμός βακτ. πληθυσμού (~1 / 106) με MIC >4 mg/mL S
 Φαινότυποι Van A, λιγότερο Van B. Μετάδοση με transposon και σε άλλα gram + Αντοχή και σε αμινογλυκοσίδες, αμπικιλλίνη. S. aureus & CNS (VRSA ή VISA) Συχνά προηγείται heteroresistance. Μικρός αριθμός βακτ. πληθυσμού (~1 / 106) με MIC >4 mg/mL. S.")
235
Κλινικές ενδείξεις γλυκοπεπτιδίων
Λοιμώξεις από MRSA ή CNS ή από PRSP Ενδοκαρδίτιδα, δέρματος, οστών, αρθρώσεων, πνευμονία, πρωτοπαθής σήψη κά. Αλλεργία στα β λακταμικά (για σταφυλόκοκκο ή πνευμονιόκοκκο) (λιγότερο αποτελεσματική από β λακταμικά) Λοιμώξεις από εντερόκοκκο Λοιμώξεις από Corynebacterium jeikeium Σταφυλοκοκκική περιτονίτιδα σε CAPD Ψευδομεμβρανώδης – βαριά- κολίτιδα (per Os).
 (λιγότερο αποτελεσματική από β λακταμικά) Λοιμώξεις από εντερόκοκκο. Λοιμώξεις από Corynebacterium jeikeium. Σταφυλοκοκκική περιτονίτιδα σε CAPD. Ψευδομεμβρανώδης – βαριά- κολίτιδα (per Os).")
236
Ανεπιθύμητες ενέργειες γλυκοπεπτιδίων
Αλλεργία (πυρετός, αναφυλακτική αντίδραση, εξάνθημα) Φλεβίτιδα (βανκο) Σύνδρομο ερυθρού αυχένα - red man syndrome- Ωτοτοξικότητα (σπανιότερα με τεϊκο) Νεφροτοξικότητα (σπάνια) Ουδετεροπενία, θρομβοπενία σε μακρά χορήγηση Καρδιαγγειακές διαταραχές ( ΑΠ ή ανακοπή, σε ταχεία iv βανκο). Auditory impairment, sometimes permanent, is associated with excessive plasma concentrations
 Φλεβίτιδα (βανκο) Σύνδρομο ερυθρού αυχένα - red man syndrome- Ωτοτοξικότητα (σπανιότερα με τεϊκο) Νεφροτοξικότητα (σπάνια) Ουδετεροπενία, θρομβοπενία σε μακρά χορήγηση. Καρδιαγγειακές διαταραχές ( ΑΠ ή ανακοπή, σε ταχεία iv βανκο). Auditory impairment, sometimes permanent, is associated with excessive plasma concentrations.")
237
Δοσολογία γλυκοπεπτιδίων
Βανκομυκίνη: 30 – 40 (-60) mg/kg/d (σε φυσιολογική νεφρική λειτουργία) Χορηγείται κάθε 12 ή κάθε 6-8 ώρες (αραίωση < 5 mg/ml , έγχυση σε >60 min) Με συνεχή έγχυση καλύτερα επίπεδα (αντιμετώπιση σοβαρών λοιμώξεων) Θεραπευτικά επίπεδα – στόχος (trough concentrations) 10 – 15 mcg/mL: βακτηριαιμία, λοιμώξεις δέρματος και μαλακών μορίων 15 – 20 mcg/mL: οστεομυελίτιδα, μηνιγγίτιδα, πνευμονία, ενδοκαρδίτιδα ΕΠΙΠΕΔΑ <10 mcg/mL ΠΡΕΠΕΙ ΝΑ ΑΠΟΦΕΥΓΟΝΤΑΙ (ΚΙΝΔΥΝΟΣ ΑΝΑΠΤΥΞΗΣ ΑΝΤΟΧΗΣ)
 mg/kg/d (σε φυσιολογική νεφρική λειτουργία) Χορηγείται κάθε 12 ή κάθε 6-8 ώρες (αραίωση < 5 mg/ml , έγχυση σε >60 min) Με συνεχή έγχυση καλύτερα επίπεδα (αντιμετώπιση σοβαρών λοιμώξεων) Θεραπευτικά επίπεδα – στόχος (trough concentrations) 10 – 15 mcg/mL: βακτηριαιμία, λοιμώξεις δέρματος και μαλακών μορίων. 15 – 20 mcg/mL: οστεομυελίτιδα, μηνιγγίτιδα, πνευμονία, ενδοκαρδίτιδα. ΕΠΙΠΕΔΑ <10 mcg/mL ΠΡΕΠΕΙ ΝΑ ΑΠΟΦΕΥΓΟΝΤΑΙ (ΚΙΝΔΥΝΟΣ ΑΝΑΠΤΥΞΗΣ ΑΝΤΟΧΗΣ)")
238
Τεϊκοπλανίνη Πλεονεκτήματα (σε σύγκριση με βανκομυκίνη) Μειονεκτήματα
Λιγότερες ανεπιθύμητες ενέργειες Χορήγηση iv ή im άπαξ ημερησίως (κατάλληλη και για εξωτερικούς ασθενείς) Σπάνια πρόκληση φλεβίτιδας Μειονεκτήματα Λιγότερα δεδομένα για ιδανική δοσολογία (?10 mg/kg) Σε βαριές λοιμώξεις πρέπει να χορηγείται σε υψηλότερη (π.χ. 12–15 mg/kg / day), από τη μέχρι προσφάτως συνιστώμενη δόση δοσολογίας σε ΝΑ (μετά 4η ημ): ½ δόσης σε Cl 40-60, 1/3 δόσης σε Cl <40 Περιορισμένη δυνατότητα μέτρησης επιπέδων (αν και είναι απαραίτητα) Μικρότερη εμπειρία για επίπεδα (trough levels: >20 mg/L). Teicoplanin may be a preferred choice if outpatient therapy is considered. It must be mentioned that less information is available regarding teicoplanin dosages and care must be taken to avoid sub-therapeutic dosing and to ensure that adequate serum concentrations are maintained. This will often require dosing well above the currently recommended guidelines (e.g. 12–15 mg/kg per day). Serum assays for teicoplanin levels are not widely available but trough levels in serum need to be measured to ensure therapeutic efficacy. Trough levels for teicoplanin probably should also be above 20 mg/L. Glycopeptide antibiotics have been evaluated and used in combination with third-generation cephalosporins, carbapenems, monobactams and quinolones. Kelsey et al. [28] compared the combination of piperacillin and gentamicin with vancomycin and aztreonam in patients with febrile neutropenia. The results in both groups were comparable.
 Σπάνια πρόκληση φλεβίτιδας. Μειονεκτήματα. Λιγότερα δεδομένα για ιδανική δοσολογία ( 10 mg/kg) Σε βαριές λοιμώξεις πρέπει να χορηγείται σε υψηλότερη (π.χ. 12–15 mg/kg / day), από τη μέχρι προσφάτως συνιστώμενη δόση. δοσολογίας σε ΝΑ (μετά 4η ημ): ½ δόσης σε Cl 40-60, 1/3 δόσης σε Cl <40. Περιορισμένη δυνατότητα μέτρησης επιπέδων (αν και είναι απαραίτητα) Μικρότερη εμπειρία για επίπεδα (trough levels: >20 mg/L). Teicoplanin may be a preferred choice if outpatient therapy is considered. It must be mentioned that. less information is available regarding teicoplanin dosages. and care must be taken to avoid sub-therapeutic dosing and. to ensure that adequate serum concentrations are maintained. This will often require dosing well above the currently recommended. guidelines (e.g. 12–15 mg/kg per day). Serum assays. for teicoplanin levels are not widely available but trough. levels in serum need to be measured to ensure therapeutic. efficacy. Trough levels for teicoplanin probably should. also be above 20 mg/L. Glycopeptide antibiotics have been. evaluated and used in combination with third-generation. cephalosporins, carbapenems, monobactams and quinolones. Kelsey et al. [28] compared the combination of piperacillin. and gentamicin with vancomycin and aztreonam in patients. with febrile neutropenia. The results in both groups were. comparable.")
239
ΛΙΝΚΟΖΑΜΙΔΕΣ ΛΙΝΚΟΜΥΚΙΝΗ ΚΛΙΝΔΑΜΥΚΙΝΗ (Dalacin)
1962, από Streptomyces lincolnensis Δεν κυκλοφορεί πλέον ΚΛΙΝΔΑΜΥΚΙΝΗ (Dalacin)
")
240
Φαρμακοκινητική κλινδαμυκίνης
Απορρόφηση: 90% Μέγιστη συγκέντρωση στο αίμα σε min Σύνδεση με λευκώματα: 84% Ημιπερίοδος ζωής: 2,5 ώρες (8-12 h σε ηπατική ανεπάρκεια) Κατανομή: σε οστά, προστάτη κ.ά. (όχι σε ΕΝΥ ή χολή σε απόφραξη) Εισέρχεται στο εσωτερικό των φαγοκυττάρων Μεταβολισμός & απέκκριση: ηπατικός (5-10% με τα ούρα) Absorption Clindamycin is nearly completely absorbed following oral administration. Food does not affect absorption significantly. The t1/2 of the antibiotic is ~3 hours. Clindamycin palmitate, an oral prodrug, is hydrolyzed rapidly in vivo. Its absorption is similar to clindamycin. The phosphate ester of clindamycin, given parenterally, also is rapidly hydrolyzed to the active parent compound. Distribution Clindamycin is widely distributed in many fluids and tissues, including bone. Significant concentrations are not attained in CSF, but concentrations sufficient to treat cerebral toxoplasmosis are achieved. The drug readily crosses the placenta. Ninety percent or more of clindamycin is bound to plasma proteins. Clindamycin accumulates in polymorphonuclear leukocytes and alveolar macrophages and in abscesses.
 Κατανομή: σε οστά, προστάτη κ.ά. (όχι σε ΕΝΥ ή χολή σε απόφραξη) Εισέρχεται στο εσωτερικό των φαγοκυττάρων. Μεταβολισμός & απέκκριση: ηπατικός (5-10% με τα ούρα) Absorption Clindamycin is nearly completely absorbed following oral administration. Food. does not affect absorption significantly. The t1/2 of the antibiotic is ~3 hours. Clindamycin palmitate, an oral prodrug, is hydrolyzed rapidly in vivo. Its absorption is similar. to clindamycin. The phosphate ester of clindamycin, given parenterally, also is rapidly hydrolyzed. to the active parent compound. Distribution Clindamycin is widely distributed in many fluids and tissues, including bone. Significant concentrations are not attained in CSF, but concentrations sufficient to treat cerebral. toxoplasmosis are achieved. The drug readily crosses the placenta. Ninety percent or more of clindamycin. is bound to plasma proteins. Clindamycin accumulates in polymorphonuclear leukocytes. and alveolar macrophages and in abscesses.")
241
Αντιμικροβιακό φάσμα κλινδαμυκίνης
Στρεπτόκοκκοι (Α, B, C, G, pneumonia, Viridans) Σταφυλόκοκκοι (ανθεκτικά στελέχη, κυρίως MRSA, CNS) Actinomyces israelii, Nocardia asteroides Propionibacterium spp. (acnes, granulosum) Clostridia spp. (ανθεκτικά 10-20%) Αναερόβια (ανθεκτικά στελέχη B. fragilis) Πρωτόζωα (Plasmodium spp, P. carinii, T. gondii, Babesia spp.)
 Σταφυλόκοκκοι (ανθεκτικά στελέχη, κυρίως MRSA, CNS) Actinomyces israelii, Nocardia asteroides. Propionibacterium spp. (acnes, granulosum) Clostridia spp. (ανθεκτικά 10-20%) Αναερόβια (ανθεκτικά στελέχη B. fragilis) Πρωτόζωα (Plasmodium spp, P. carinii, T. gondii, Babesia spp.)")
242
Aνεπιθύμητες ενέργειες κλινδαμυκίνης
Διάρροια & ψευδομεμβρανώδης κολίτιδα (2-20%) Θεραπεία με μετρονιδαζόλη (PO ή IV). Σε βαριά κατάσταση Vancomycin per Os. Αλλεργικές αντιδράσεις (10%) Τρανασαμινασαιμία Λευκοπενία, θρομβοπενία Καρδιαγγειακές διαταραχές (υπόταση ή ανακοπή σε ταχεία ΕΦ χορήγηση) Θρομβοφλεβίτιδα, τοπικός ερεθισμός, άσηπτο απόστημα (im),.
 Θεραπεία με μετρονιδαζόλη (PO ή IV). Σε βαριά κατάσταση Vancomycin per Os. Αλλεργικές αντιδράσεις (10%) Τρανασαμινασαιμία. Λευκοπενία, θρομβοπενία. Καρδιαγγειακές διαταραχές (υπόταση ή ανακοπή σε ταχεία ΕΦ χορήγηση) Θρομβοφλεβίτιδα, τοπικός ερεθισμός, άσηπτο απόστημα (im),.")
243
Kλινικές ενδείξεις κλινδαμυκίνης
Ενδοκοιλιακές λοιμώξεις (με β λακταμικό ή αμινογλυκοσίδη) Πυελικές λοιμώξεις Πνευμονία από εισρόφηση, πν. απόστημα, εμπύημα Λοιμώξεις αν. αναπνευστικού; (χρόνιες) Λοιμώξεις δέρματος, μαλακών μορίων και οστών Τοπική ή συστηματική θεραπεία ακμής Εγκεφαλική τοξοπλάσμωση (με πυριμεθαμίνη) Πνευμονία από P. carinii (με πριμακίνη).
 Πυελικές λοιμώξεις. Πνευμονία από εισρόφηση, πν. απόστημα, εμπύημα. Λοιμώξεις αν. αναπνευστικού; (χρόνιες) Λοιμώξεις δέρματος, μαλακών μορίων και οστών. Τοπική ή συστηματική θεραπεία ακμής. Εγκεφαλική τοξοπλάσμωση (με πυριμεθαμίνη) Πνευμονία από P. carinii (με πριμακίνη).")
244
Δοσολογία κλινδαμυκίνης
Ενήλικες Από στόμα: mg / 6 ώρες (μέχρι mg / 6 h). Ενδοφλεβίως: mg κάθε 6-8 ώρες (βαριές λοιμώξεις) Παιδιά: Από το στόμα: 8-16 mg/kg (μέχρι mg/kg/d) Ενδοφλεβίως: mg/kg/d Σε ηπατική ανεπάρκεια: 1/2 της δόσης
. Ενδοφλεβίως: mg κάθε 6-8 ώρες (βαριές λοιμώξεις) Παιδιά: Από το στόμα: 8-16 mg/kg (μέχρι mg/kg/d) Ενδοφλεβίως: mg/kg/d. Σε ηπατική ανεπάρκεια: 1/2 της δόσης.")
245
Ανταγωνιστές φυλικού οξέος
Σουλφοναμίδη Τριμεθοπρίμη ΠΑΒΟ Διϋδροφυλλικό Τετραϋδροφυλλικό οξύ διϋδροπτεροϊκή συνθετάση διϋδροφυλλική αναγωγάση Τα βακτήρια πρέπει να συνθέσουν φυλικό οξύ (δεν το προσλαμβάνουν έτοιμο)
")
246
Σουλφοναμίδες Δυσαπορρόφητες Παρατεταμένης δράσης Βραχείας δράσης
Παράγωγα σουλφανυλουρίας Σουλφαδιμεδίνη Σουλφακεταμίδη Μέσης δράσης Σουλφαδιαζίνη Σουλφαμεθοξαζόλη Μακράς δράσης Σουλφαμεθοξυδιαζίνη Παρατεταμένης δράσης Σουλφαμετοπυραζίνη Σουλφαδοξίνη Δυσαπορρόφητες Φορμοφθαλυλο-σουλφακαρβαναμίδη Σουλφαγουανιδίνη Σουλφαθαλιδίνη Σήμερα χρησιμοποιείται σχεδόν αποκλειστικά η Sulfamethoxazole σε συνδυασμό (5:1) με Trimethoprim (co-trimoxazole, TMP-sulfa, TMP-SMX) λόγω συνέργειας
 με Trimethoprim (co-trimoxazole, TMP-sulfa, TMP-SMX) λόγω συνέργειας.")
247
Αντιμικροβιακό φάσμα Co-Trimoxazol
Gram θετικοί κόκκοι Στρεπτόκοκκοι (S. pyogenes, S. pneumonia, S. viridans) Enterococcus spp (ανθεκτικά στελέχη) Staphylococcus spp. (ανθεκτικά στελέχη) Gram αρνητικοί κόκκοι Neisseria gonorrhoeae, N. meningitides Gram θετικοί βάκιλλοι Νοκάρδια αστεροειδής Gram αρνητικοί βάκιλλοι (ανθεκτικά στελέχη) Y. enterocolitica, Ε. coli, P. mirabilis, Salmonella spp, Shigella spp, Klebsiella spp, Serratia spp, Enterobacter, Citrobacter, V. cholera , H. influenzae, Brucella spp. Πρωτόζωα (μικροσπορίδια, P. carinii, Isospora belli)
 Enterococcus spp (ανθεκτικά στελέχη) Staphylococcus spp. (ανθεκτικά στελέχη) Gram αρνητικοί κόκκοι. Neisseria gonorrhoeae, N. meningitides. Gram θετικοί βάκιλλοι. Νοκάρδια αστεροειδής. Gram αρνητικοί βάκιλλοι (ανθεκτικά στελέχη) Y. enterocolitica, Ε. coli, P. mirabilis, Salmonella spp, Shigella spp, Klebsiella spp, Serratia spp, Enterobacter, Citrobacter, V. cholera , H. influenzae, Brucella spp. Πρωτόζωα (μικροσπορίδια, P. carinii, Isospora belli)")
248
Φαρμακοκινητική Co-Trimoxazol
ΤΜΠ ΣΜΞ Απορρόφηση σχεδόν πλήρης σχεδόν πλήρης Σύνδεση με λεύκωμα % 70% Ημιπερίοδος ζωής ώρες 10 ώρες Απέκκριση % % (ούρα 24ώρου) Κατανομή Πολύ καλή: νεφροί, πνεύμονες. Σχετικώς καλή: σίελος, βρογχικές εκκρίσεις, υδατοειδές υγρό, χολή, προστάτης Μικρού βαθμού: ΕΝΥ.
 Κατανομή. Πολύ καλή: νεφροί, πνεύμονες. Σχετικώς καλή: σίελος, βρογχικές εκκρίσεις, υδατοειδές υγρό, χολή, προστάτης. Μικρού βαθμού: ΕΝΥ.")
249
Ανεπιθύμητες ενέργειες Co-Trimoxazol
Από ΓΕΣ Ναυτία, έμετοι, διάρροια, ψευδομ.κολίτιδα Από αιμοποιητικό Μεγαλοβλαστική αναιμία, λευκοπενία, θρομβοπενία Αλλεργικές εκδηλώσεις Κνησμός, εξάνθημα, πυρετός, σ, Stevens-Johnson, σ. Lyell Αύξηση ηπατικών ενζύμων Αύξηση ουρίας, κρεατινίνης.

250
Κλινικές ενδείξεις Co-Trimoxazol
Λοιμώξεις ουροποιητικού Λοιμώξεις αν. αναπνευστικού S. pneumonia, H. influenzae, Moraxella catarrhalis Έξαρση χρόνιας βρογχίτιδας Πνευμονία από Pneumocystis jirovecii (PcP) Τυφοειδής πυρετός Εντερικές λοιμώξεις Bρουκέλλωση Νοκαρδίωση Κοκκιωμάτωση Wegener.
 Τυφοειδής πυρετός. Εντερικές λοιμώξεις. Bρουκέλλωση. Νοκαρδίωση. Κοκκιωμάτωση Wegener.")
251
Αντενδείξεις τριμεθοπρίμης - σουλφαμεθοξαζόλης
Αντενδείξεις τριμεθοπρίμης - σουλφαμεθοξαζόλης Μεγαλοβλαστική αναιμία από έλλειψη φυλλικού οξέος Βαριά ηπατική βλάβη ή οξεία ηπατίτιδα Αιματολογική δυσκρασία Κύηση Θηλασμός.

252
Δοσολογία Co-Trimoxazol
Οξείες λοιμώξεις 160 mg +800 mg κάθε 12 ώρες Οξείες βαριές λοιμώξεις 240 mg mg κάθε 12 ώρες Μακροχρόνια χορήγηση 80 mg mg κάθε 12 ώρες Πνευμονία από P. jiroveci (carini) 5 mg/kg +20 mg/kg κάθε 6 ώρες 1 απλό δισκίο ή 1 amp: 80 mg TMP mg SMX
 5 mg/kg +20 mg/kg κάθε 6 ώρες. 1 απλό δισκίο ή 1 amp: 80 mg TMP mg SMX.")
253
ΝΙΤΡΟΪΜΙΔΑΖΟΛΕΣ Μετρονιδαζόλη Flagyl, Colpocin T Ορνιδαζόλη Betiral
Τινιδαζόλη Fasigyn, Trichogin Νιμοραζόλη Naxogin Καρνιδαζόλη Σεκνιδαζόλη

254
Αντιμικροβιακό φάσμα μετρονιδαζόλης
Πρωτόζωα Entamoeba histolytica, Trichomonas vaginalis, Giardia lamblia Αναερόβια μικρόβια (εξαιρούνται: προπιονικό βακτηρίδιο, μικρoαερόφιλοι κόκκοι, ακτινομύκητες) Gardnerella vaginalis Helicobacter pylori.
 Gardnerella vaginalis. Helicobacter pylori.")
255
Φαρμακοκινητική μετρονιδαζόλης
Απορρόφηση από πεπτικό: πλήρης καλή απορρόφηση (υπόθετο) από ορθό ή κόλπο Ημιπερίοδος ζωής: 7-8 ώρες Σύνδεση με λευκώματα: 15%. Μεταβολισμός: ηπατικός απέκκριση από νεφρούς (δεν απαιτείται προσαρμογή) Κατανομή: Σχεδόν παντού εγκέφαλο, ήπαρ, μήτρα, λίπος, δέρμα, οστά, αποστηματικές κοιλότητες, ΕΝΥ, σίελο, περιτοναϊκό υγρό, εκκρίσεις κόλπου, γάλα κ.ά.
 από ορθό ή κόλπο. Ημιπερίοδος ζωής: 7-8 ώρες. Σύνδεση με λευκώματα: 15%. Μεταβολισμός: ηπατικός. απέκκριση από νεφρούς (δεν απαιτείται προσαρμογή) Κατανομή: Σχεδόν παντού εγκέφαλο, ήπαρ, μήτρα, λίπος, δέρμα, οστά, αποστηματικές κοιλότητες, ΕΝΥ, σίελο, περιτοναϊκό υγρό, εκκρίσεις κόλπου, γάλα κ.ά.")
256
Ανεπιθύμητες ενέργειες μετρονιδαζόλης
Γαστρεντερικές διαταραχές (3%) Ναυτία, έμετοι, επιγαστρική δυσφορία, ανορεξία, μεταλλική γεύση, Διαταραχές από νευρικό σύστημα Περιφ. νευροπάθεια, σπασμοί, αταξία, σύγχυση (σε υψηλή δοσολογία) Δυσανεξία στο αλκοόλ (σ. δισουλφιράμης) Ουδετεροπενία, θρομβοπενία Αλλεργικές αντιδράσεις (σπάνια) Ανάπτυξη μυκητιάσεων Καρκινογένεση σε πειραματόζωα GI Abdominal discomfort Metallic taste Furring of tongue and glossitis Acute pancreatitis Hypersensitivity Rashes, pruritus, bronchospasm CNS Peripheral neuropathy, ataxia, confusion, seizures Higher doses
 Ναυτία, έμετοι, επιγαστρική δυσφορία, ανορεξία, μεταλλική γεύση, Διαταραχές από νευρικό σύστημα. Περιφ. νευροπάθεια, σπασμοί, αταξία, σύγχυση (σε υψηλή δοσολογία) Δυσανεξία στο αλκοόλ (σ. δισουλφιράμης) Ουδετεροπενία, θρομβοπενία. Αλλεργικές αντιδράσεις (σπάνια) Ανάπτυξη μυκητιάσεων. Καρκινογένεση σε πειραματόζωα. GI. Abdominal discomfort. Metallic taste. Furring of tongue and glossitis. Acute pancreatitis. Hypersensitivity. Rashes, pruritus, bronchospasm. CNS. Peripheral neuropathy, ataxia, confusion, seizures. Higher doses.")
257
Κλινικές ενδείξεις μετρονιδαζόλης
Ενδοκοιλιακές και πυελικές λοιμώξεις Απόστημα εγκεφάλου ή μηνιγγίτιδα από αναερόβια Ενδομητρίτιδα, επιλόχεια λοίμωξη Κολπίτιδα από Trichomonas vaginalis ή Gardnerella vaginalis Ψευδομεμβρανώδης κολίτιδα από C. difficile Διάρροια από υπερανάπτυξη μικροβίων Νόσος Crohn Λοιμώξεις από πρωτόζωα Giardia lamblia, Entamoeba histolytica Vaginitis due to Trichomonas vaginalis (protozoal) infection in both symptomatic patients as well as their asymptomatic sexual contacts; Pelvic inflammatory disease in conjunction with other antibiotics such as ofloxacin, levofloxacin, or ceftriaxone Protozoal infections due to Entamoeba histolytica (Amoebic dysentery or Hepatic abscesses), and Giardia lamblia (Giardiasis) should be treated alone or in conjunction with iodoquinol or diloxanide furoate Anaerobic bacterial infections such as Bacteroides fragilis, spp, Fusobacterium spp, Clostridium spp, Peptostreptococcus spp, Prevotella spp, or any other anaerobes in intraabdominal abscess, peritonitis, empyema, pneumonia, aspiration pneumonia, lung abscess, diabetic foot ulcer, meningitis and brain abscess, bone and joint infections, septicemia, endometritis, tubo-ovarian abscess, or endocarditis Pseudomembranous colitis due to Clostridium difficile Helicobacter pylori eradication therapy, as part of a multi-drug regimen in peptic ulcer disease Prophylaxis for those undergoing potentially contaminated colorectal surgery and may be combined with neomycin
 infection in both symptomatic patients as well as their asymptomatic sexual contacts; Pelvic inflammatory disease in conjunction with other antibiotics such as ofloxacin, levofloxacin, or ceftriaxone. Protozoal infections due to Entamoeba histolytica (Amoebic dysentery or Hepatic abscesses), and Giardia lamblia (Giardiasis) should be treated alone or in conjunction with iodoquinol or diloxanide furoate. Anaerobic bacterial infections such as Bacteroides fragilis, spp, Fusobacterium spp, Clostridium spp, Peptostreptococcus spp, Prevotella spp, or any other anaerobes in intraabdominal abscess, peritonitis, empyema, pneumonia, aspiration pneumonia, lung abscess, diabetic foot ulcer, meningitis and brain abscess, bone and joint infections, septicemia, endometritis, tubo-ovarian abscess, or endocarditis. Pseudomembranous colitis due to Clostridium difficile. Helicobacter pylori eradication therapy, as part of a multi-drug regimen in peptic ulcer disease. Prophylaxis for those undergoing potentially contaminated colorectal surgery and may be combined with neomycin.")
258
Ριφαμυκίνες Rifampin Rifabutin Rifaximin (Rifacol tbl 200 mg)
Παλαιότερη και συχνότερα χρησιμοποιούμενη ριφαμυκίνη Ισχυρός επαγωγέας CY P450 Μεταβολίζεται στο ήπαρ Έχει πολύ καλή δραστικότητα σε biofilm Rifabutin Επάγει λιγότερο το CY P450 Χρησιμοποιείται σε ασθενείς που λαμβάνουν HAART Rifaximin (Rifacol tbl 200 mg) Απορροφάται ελάχιστα Χρησιμοποιείται για θεραπεία διάρροιας ταξιδιωτών και ηπατικής εγκεφαλοπάθειας
 Απορροφάται ελάχιστα. Χρησιμοποιείται για θεραπεία διάρροιας ταξιδιωτών. και ηπατικής εγκεφαλοπάθειας.")
259
Αντιμικροβιακό φάσμα ριφαμπικίνης
Gram-θετικά Σταφυλόκοκκοι Gram-αρνητικά Haemophilus influenzae, Neisseria meningitidis Μυκοβακτηρίδια Mycobacterium tuberculosis, Mycobacterium avium complex, Mycobacterium leprae.

260
Κλινικές ενδείξεις ριφαμπικίνης
Φυματίωση και λοίμωξη από μη φυματιώδη μυκοβακτηρίδια Σε συνδυασμό με άλλα αντιφυματικά φάρμακα Θεραπεία σταφυλοκοκκικών λοιμώξεων Σε συνδυασμό με άλλα αντισταφυλοκοκκικά Πρόληψη μηνιγγίτιδας από μηνιγγιτιδόκοκκο ή αιμόφιλο

261
Κλινικές ενδείξεις ριφαμυκίνης
Ηπατική εγκεφαλοπάθεια (1200 mg/8h μέχρι 7 ημέρες) Διάρροια ταξιδιωτών και λοιμώδης γαστρεντερίτιδα (800 mg/8h μέχρι 7 ημέρες) Αντισηψία εντέρου σε χειρουργικές επεμβάσεις
 Διάρροια ταξιδιωτών και λοιμώδης γαστρεντερίτιδα (800 mg/8h μέχρι 7 ημέρες) Αντισηψία εντέρου σε χειρουργικές επεμβάσεις.")
262
Νέα αντιβιοτικά για MRSA
Linezolid Daptomycin Quinupristin-dalfopristin

263
Linezolid (Zyvoxid) Ανήκει στις οξαζολιδόνες (Oxazolidinone)
Αναστέλλει την πρωτεϊνική σύνθεση βακτηρίων σύνδεση με 50S υπομονάδα ριβοσωματίων (βακτηριοστατικό) Διατίθεται σε IV και PO μορφή (ισοδύναμες) Ενδέχεται να υπερτερεί vancomycin για πνευμονία από MRSA .
 Διατίθεται σε IV και PO μορφή (ισοδύναμες) Ενδέχεται να υπερτερεί vancomycin για πνευμονία από MRSA. .")
264
Αντιμικροβιακό φάσμα Linezolid
Streptococcus pyogenes S. Viridans S. pneumoniae (+PRSP) Staphylococcuw sp. (+VRSA) Enterococcus sp. (+VRE)
 Staphylococcuw sp. (+VRSA) Enterococcus sp. (+VRE)")
265
Φαρμακοκινητική Linezolid
Απορρόφηση 100% (ανεξαρτήτως τροφής) Καλή διείσδυση σε πνεύμονες και άλλους ιστούς (ΕΝΥ 70%) Απέκκριση – νεφρική και ηπατική t½ 4-6 h δεν απαιτείται προσαρμογή σε νεφρική ανεπάρκεια Σύνδεση με λευκώματα: 30% Δοσολογία (IV ή PO): 600 mg Q12H
 Καλή διείσδυση σε πνεύμονες και άλλους ιστούς (ΕΝΥ 70%) Απέκκριση – νεφρική και ηπατική. t½ 4-6 h. δεν απαιτείται προσαρμογή σε νεφρική ανεπάρκεια. Σύνδεση με λευκώματα: 30% Δοσολογία (IV ή PO): 600 mg Q12H.")
266
ΚΛΙΝΙΚΕΣ ΕΝΔΕΙΞΕΙΣ Λοιμώξεις από vancomycin-resistant E. faecium
Νοσοκομειακή πνευμονία από MSSA & MRSA Πνευμονιοκοκκική πνευμονία κοινότητας Επιπλεγμένη λοίμωξη δέρματος και μαλακών μορίων από ανθεκτούς S. aureus

267
Ανεπιθύμητες ενέργειες Linezolid
Νευροτοξικότητα, μυελοκαταστολή σε >2 εβδ χορήγηση (2 - 4%) Γαστρεντερικές διαταραχές: ναυτία, έμετοι, διάρροια (6 - 8 %) Κεφαλαλγία (6.5%) Εξάνθημα Αλληλεπιδράσεις: Μη εκλεκτικοί ασθενείς αναστολείς MAO Αντικαταθλιπτικά (< 1%) Πλούσιες σε τυραμίνη τροφές (τυρί, μπύρα, κρασί, καπνιστά κρέατα κ.ά) Αυξημένο κόστος
 Γαστρεντερικές διαταραχές: ναυτία, έμετοι, διάρροια (6 - 8 %) Κεφαλαλγία (6.5%) Εξάνθημα. Αλληλεπιδράσεις: Μη εκλεκτικοί ασθενείς αναστολείς MAO. Αντικαταθλιπτικά (< 1%) Πλούσιες σε τυραμίνη τροφές (τυρί, μπύρα, κρασί, καπνιστά κρέατα κ.ά) Αυξημένο κόστος.")
268
Daptomycin (Cubicin) Νέο κυκλικό λιποπεπτίδιο (2007)
Βακτηριοκτόνο, concentration-dependent, μετα-αντιβιοτική δράση Συνδέεται στη βακτηριακή μεμβράνη (παρουσία Ca2+) - ταχεία εκπόλωση κυτταρικός θάνατος
 - ταχεία εκπόλωση κυτταρικός θάνατος.")
269
Φαρμακοκινητική και δόση Daptomycin
Κατανομή: καλή διείσδυση σε ιστούς t/2: 8-9 h (έως 28 h σε νεφρική ανεπάρκεια) Αποβολή: Ούρα (78%, αναλλοίωτο), κόπρανα (6%) Δόση: SSTIs: 4 mg/kg IV ημερησίως Βακτηριαιμία - ενδοκαρδίτιδα: 6 mg/kg IV ημερησίως Εάν CrCl < 30 κάθε 48 ώρες Μπορεί να δώσει ψευδώς υψηλό INR
 Αποβολή: Ούρα (78%, αναλλοίωτο), κόπρανα (6%) Δόση: SSTIs: 4 mg/kg IV ημερησίως. Βακτηριαιμία - ενδοκαρδίτιδα: 6 mg/kg IV ημερησίως. Εάν CrCl < 30 κάθε 48 ώρες. Μπορεί να δώσει ψευδώς υψηλό INR.")
270
Αντιμικροβιακό φάσμα Daptomycin
Gram-θετικοί κόκκοι Streptococcus pyogenes S. viridans S. pneumoniae Staphylococcus spp. Enterococcus spp. Clostridium spp. Daptomycin MIC Values (µg/mL) Organism N Range MIC50 MIC90 E. faecalis (VR-strains) 34 0.25 – 2 0.5 2 E. faecalis (VS-strains) 917 ≤0.06 – 4 1 E. faecium (incl. VR-strains) 398 0.25 – 4 4 S. epidermidis (incl. MR-strains) 164 0.12 – 1 CUBICIN® Product Monograph September 2007.
 Organism. N. Range. MIC50. MIC90. E. faecalis (VR-strains) – E. faecalis (VS-strains) 917. ≤0.06 – E. faecium (incl. VR-strains) – S. epidermidis (incl. MR-strains) – 1. CUBICIN® Product Monograph September")
271
Κλινικές ενδείξεις Daptomycin
Επιπλεγμένες λοιμώξεις δέρματος και μαλακών μορίων Βακτηριαιμία από Staphylococcus και ενδοκαρδίτιδα δεξιών κοιλοτήτων Δεν είναι αποτελεσματική σε πνευμονία λόγω σύνδεσης και αδρανοποίησης από surfactant. Gram positive bacteria includes: mssa, mrsa, streptococcus sp., e. faecalis (vanc susceptible) Mechanism of action: Insertion of lipophilic tail into the cell membrane Daptomycin oligomerizes disrupts membrane Membrane depolarization, potassium ion efflux - this results in arrest of dna, rna, and protein synthesis cell death Daptomycin resistant isolates: would have increased voltage difference across the cytoplasmic membrane Susceptibilities: < 1 mcg/mL for staphylococci and strep; < 4 mcg/mL for enterococcus
 Mechanism of action: Insertion of lipophilic tail into the cell membrane. Daptomycin oligomerizes disrupts membrane. Membrane depolarization, potassium ion efflux - this results in arrest of dna, rna, and protein synthesis cell death. Daptomycin resistant isolates: would have increased voltage difference across the cytoplasmic membrane. Susceptibilities: < 1 mcg/mL for staphylococci and strep; < 4 mcg/mL for enterococcus.")
272
Ανεπιθύμητες ενέργειες Daptomycin
Αιματολογικές: Αναιμία Γαστρεντερικές: Διάρροια, έμετοι Αύξηση CPK

273
Quinupristin-dalfopristin (Synercid)
Σύνθεση Quinupristin – streptogramin A Dalfopristin – streptogramin B Μηχανισμός δράσης Σύνδεση με 50S ριβοσωματική υπομονάδα. Φάσμα MRSA, VISA, coag neg. staph., PRSP, VRE (όχι Enterococcus faecium) Συνέργεια με β λακταμικά Ανεπιθύμητες ενέργειες Αρθραλγίες, μυαλαγίες, υπερχολερυθριναιμία Φλεβίτιδα (απαιτεί ΚΦΚ).
 Συνέργεια με β λακταμικά. Ανεπιθύμητες ενέργειες. Αρθραλγίες, μυαλαγίες, υπερχολερυθριναιμία. Φλεβίτιδα (απαιτεί ΚΦΚ).")
274
Colistin Παλαιό, “ξεχασμένο” αντιβιοτικό λόγω τοξικότητας
Ανήκει στις πολυμιξίνες (1947, από είδη Bacillus polymyxa) Αυξημένος κίνδυνος νευροτοξικότητας, νεφροτοξικότητας Χρησιμοποιείται σήμερα με αυξανόμενη συχνότητα για παν-ανθεκτικά Gram-αρν. Δραστικό φάσμα (απαιτείται αντιβιόγραμμα!) Pseudomonas aeruginosa Acinetobacter spp. E. coli, Klebsiella, Enterobacter, Citrobacter, Hemophilus spp. Συνέργεια με ριφαμπικίνη in vitro έναντι στελεχών Stenotrophomonas maltophilia
 Αυξημένος κίνδυνος νευροτοξικότητας, νεφροτοξικότητας. Χρησιμοποιείται σήμερα με αυξανόμενη συχνότητα για παν-ανθεκτικά Gram-αρν. Δραστικό φάσμα (απαιτείται αντιβιόγραμμα!) Pseudomonas aeruginosa. Acinetobacter spp. E. coli, Klebsiella, Enterobacter, Citrobacter, Hemophilus spp. Συνέργεια με ριφαμπικίνη in vitro έναντι στελεχών Stenotrophomonas maltophilia.")

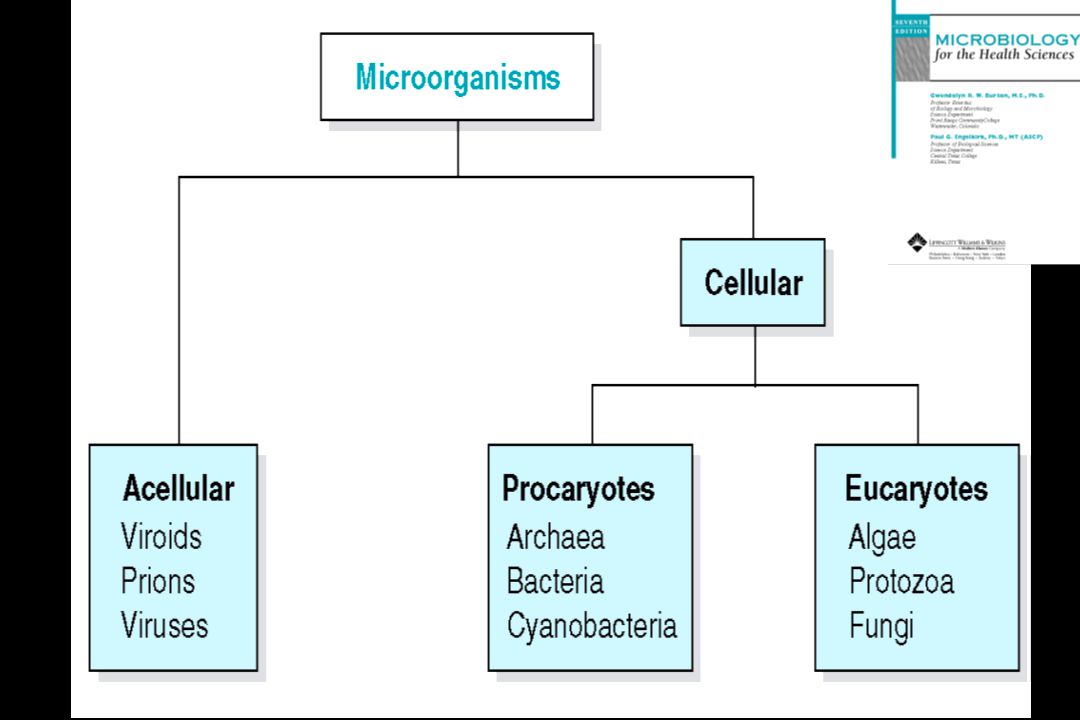
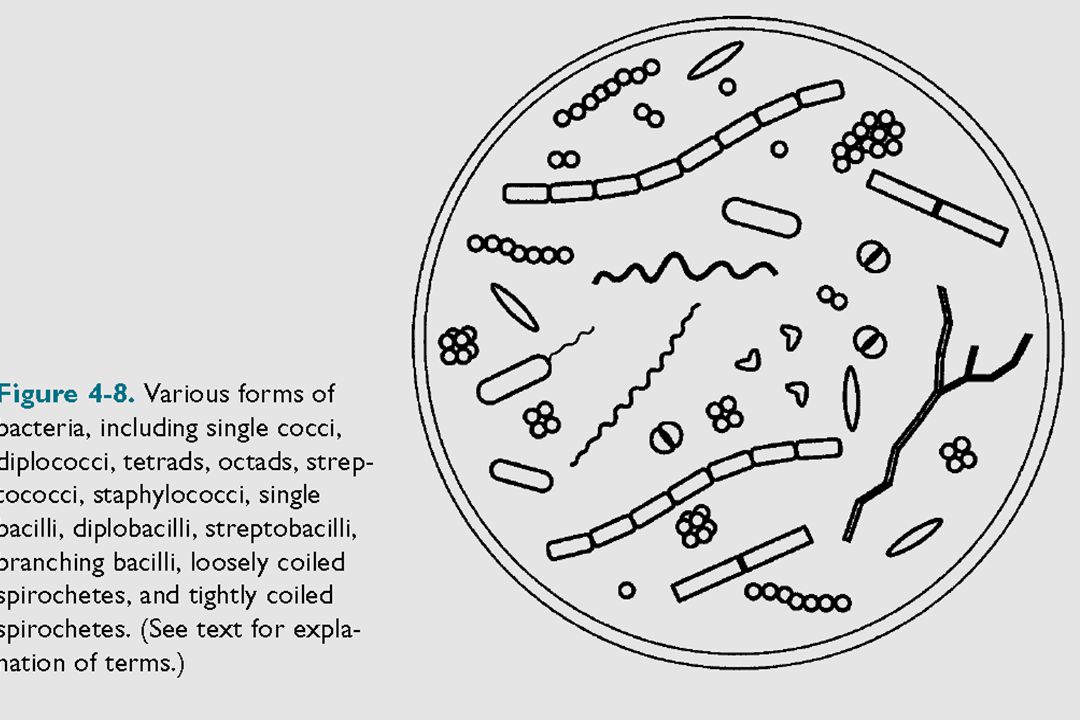
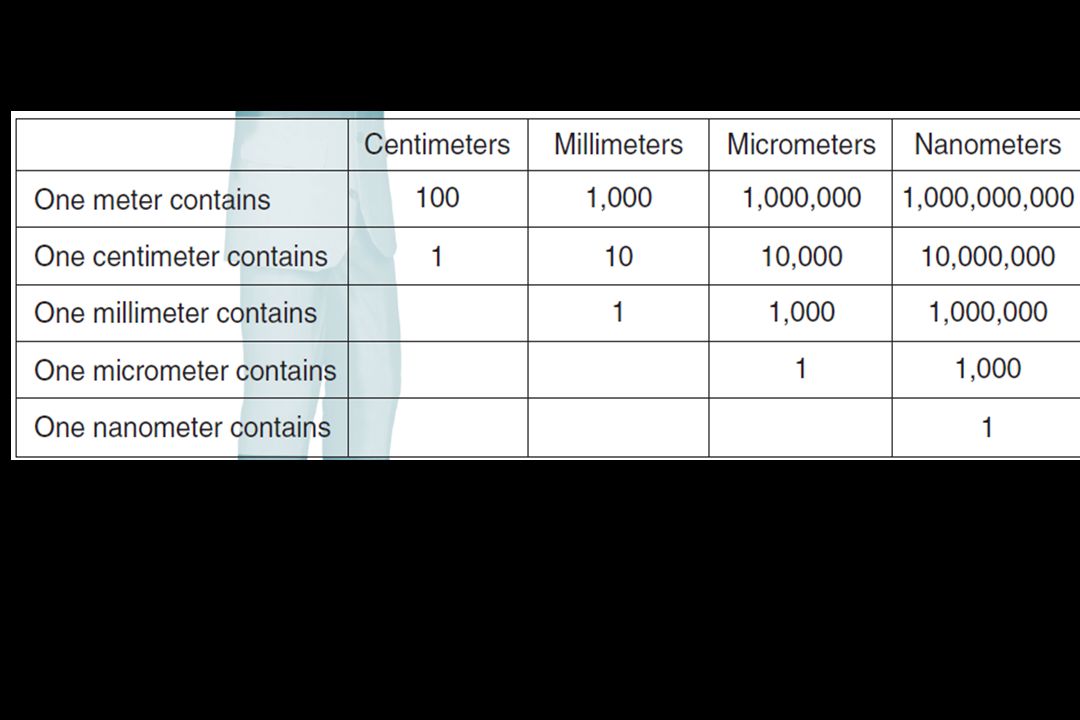


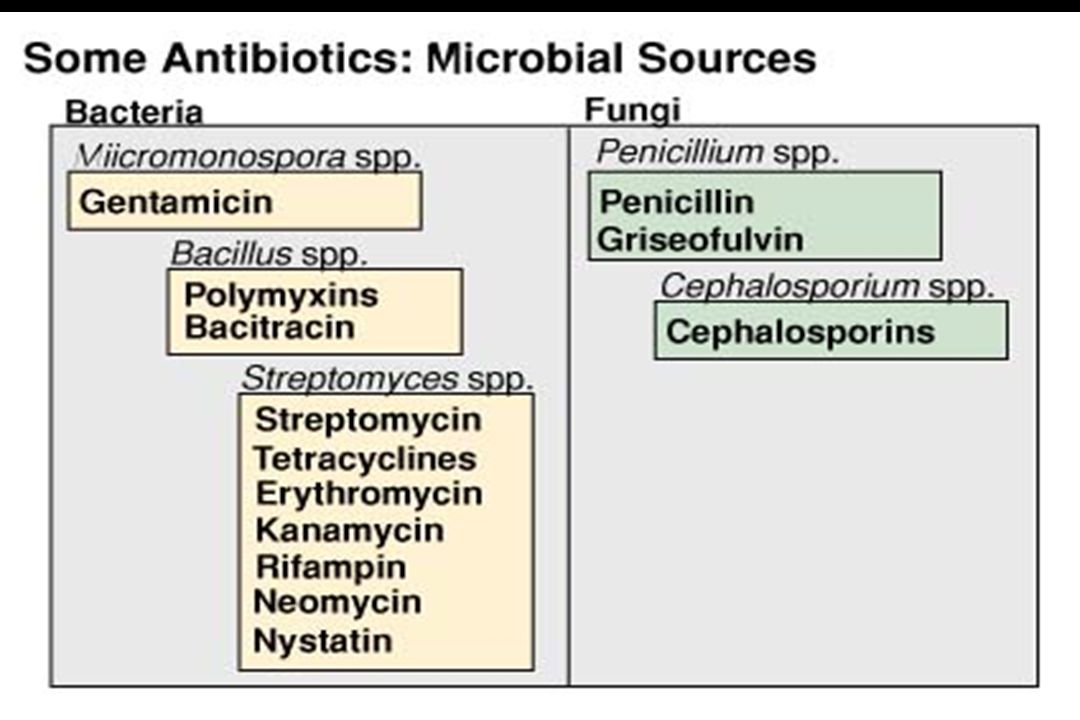



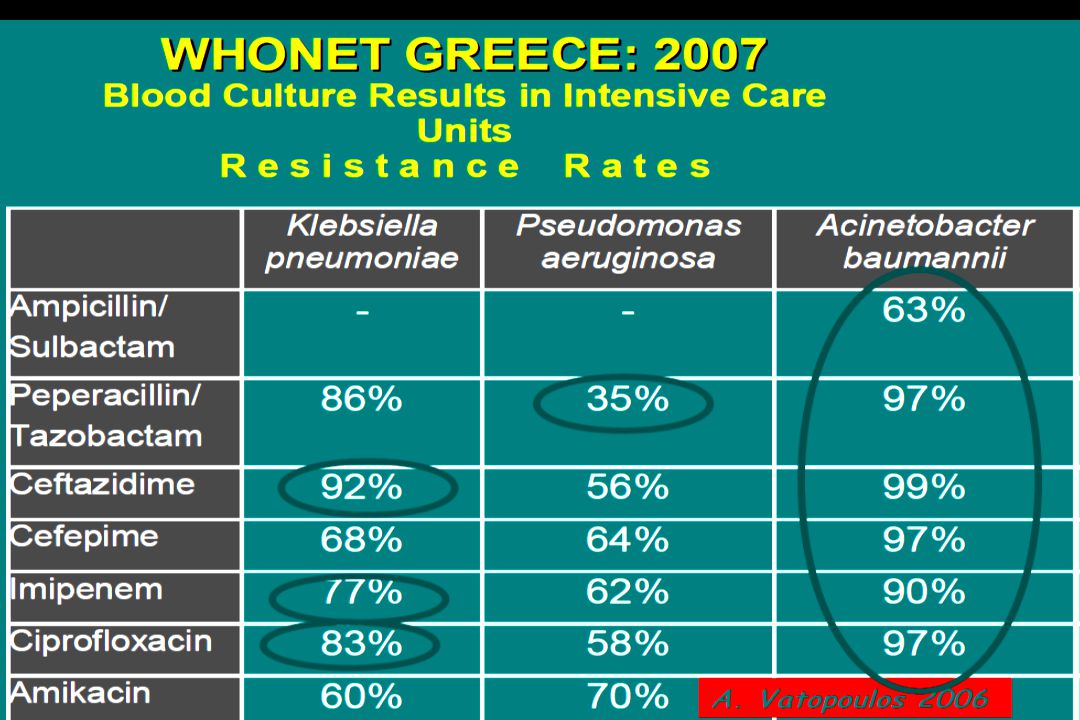
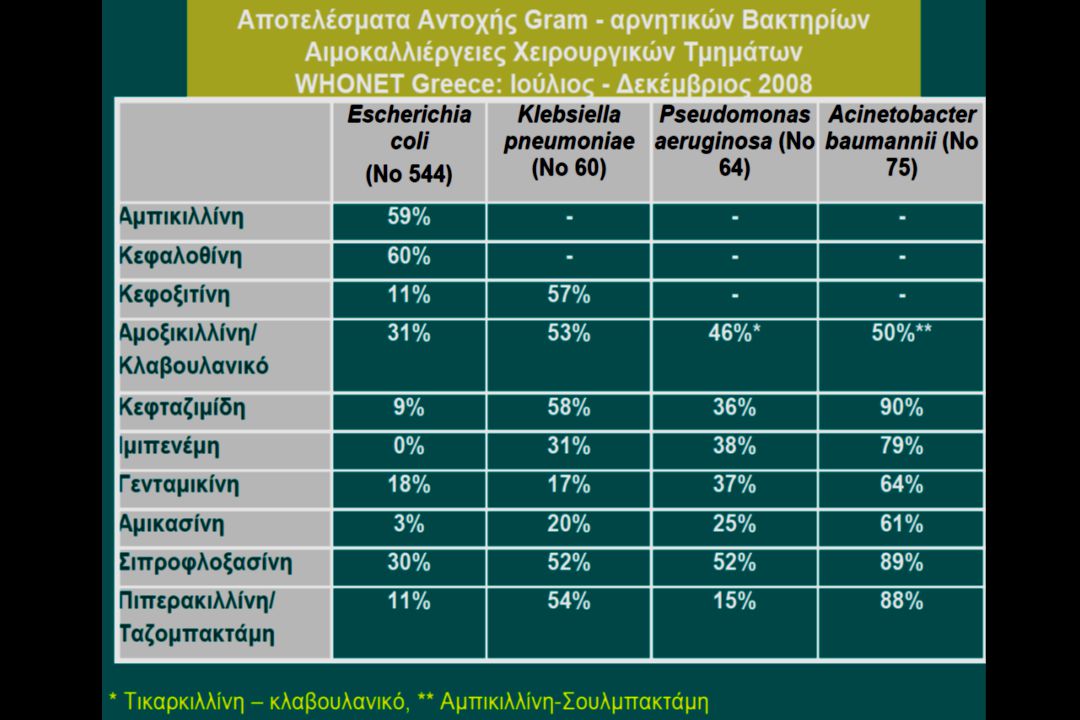
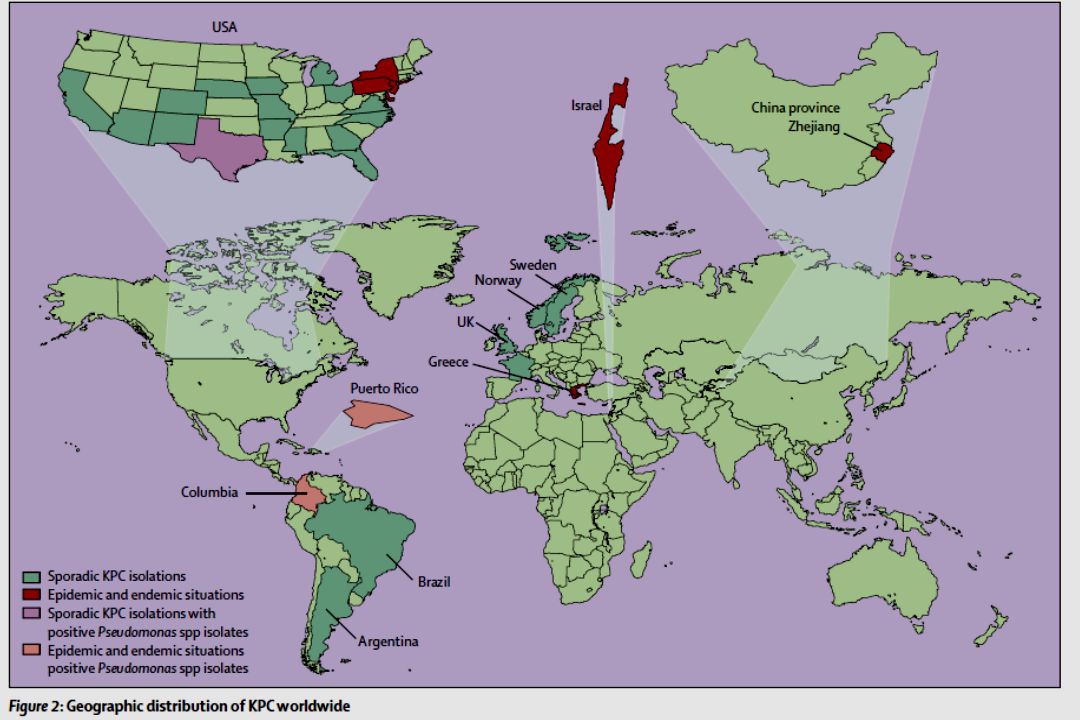





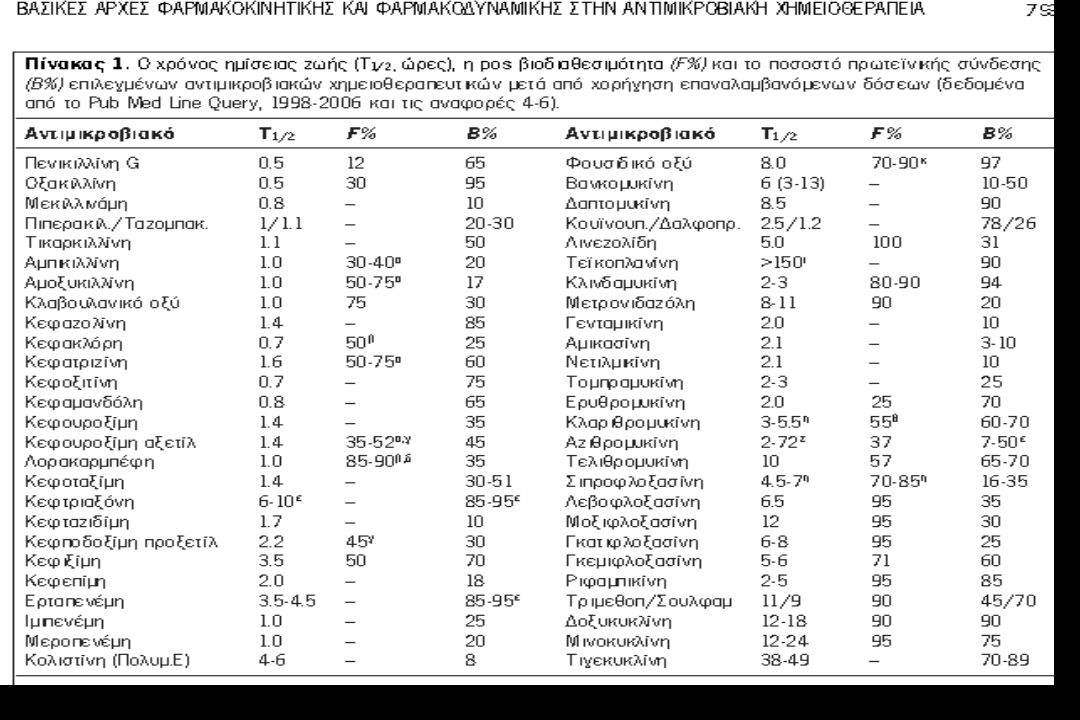
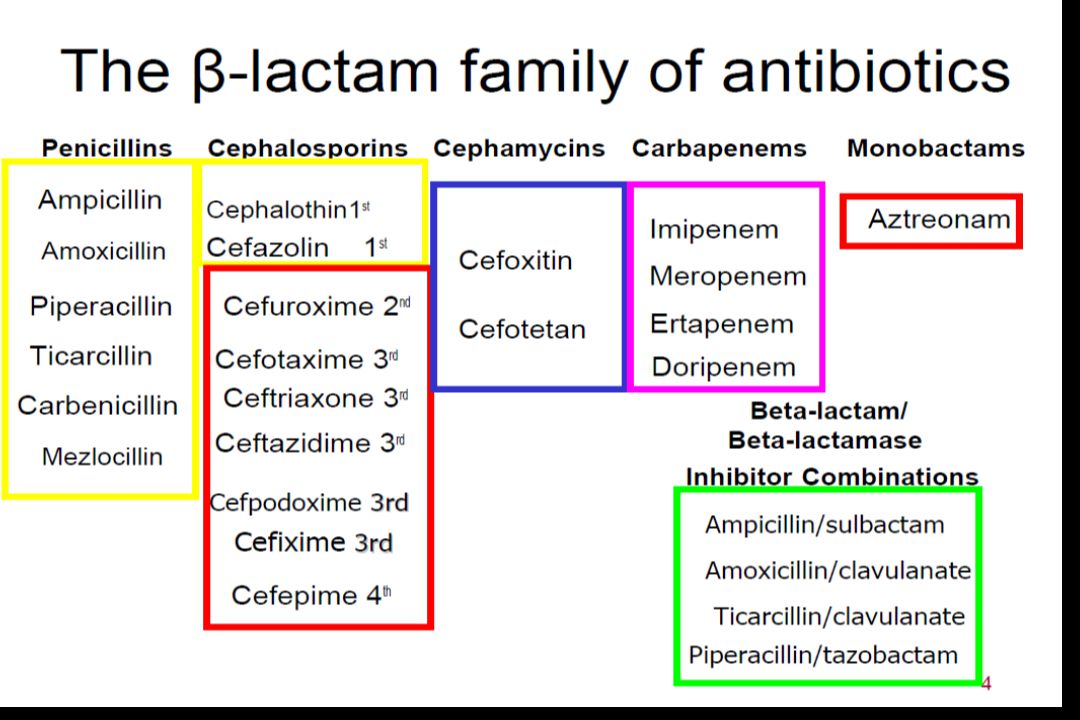
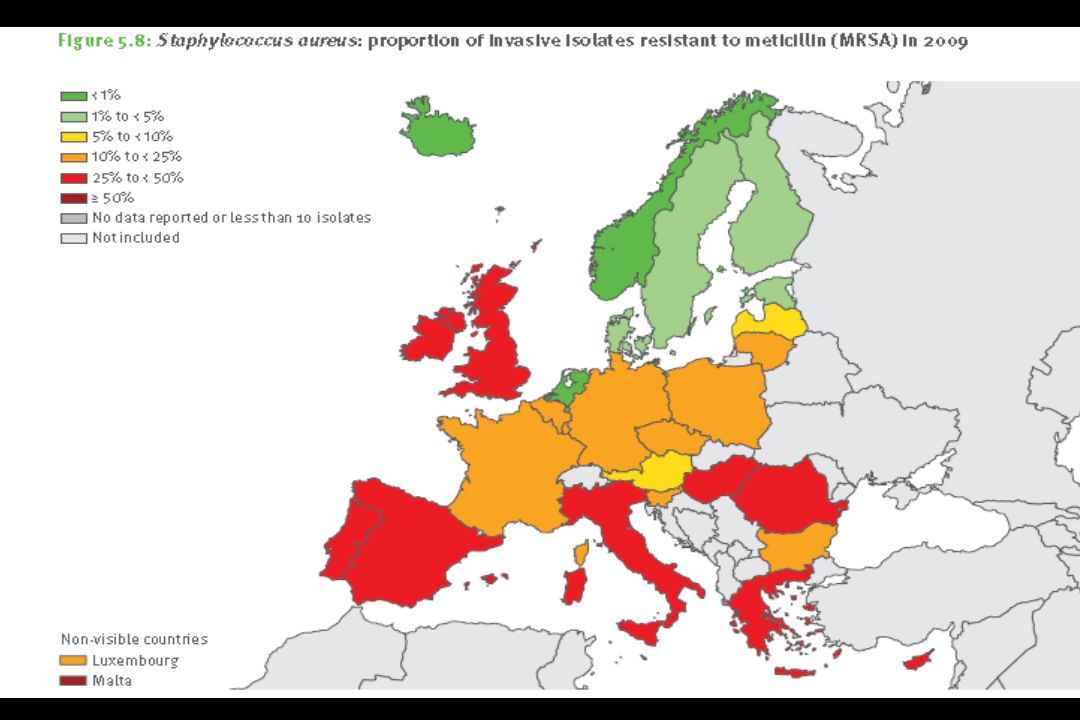






 2014>")


>")













