Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
Κληρονομούμενα σύνδρομα μυελικής ανεπάρκειας στα παιδιά Κόσσυβα Λυδία Παιδίατρος, Επ. Καθηγήτρια Παιδιατρικής Αιματολογίας, Β΄ΠΠΚ Νοσ. Παίδων «Π&Α Κυριακού» Κατ’επιλογή μάθημα Αιματολογίας-Ογκολογίας, Νοέμβριος 2014

2
Ορισμός Ετερογενής ομάδα νοσημάτων που χαρκτηρίζεται από προïούσα μυελική ανεπάρκεια σε συνδυασμό με σωματικές ανωμαλίες Ετερογενής ομάδα νοσημάτων που χαρκτηρίζεται από προïούσα μυελική ανεπάρκεια σε συνδυασμό με σωματικές ανωμαλίες

3
Σύνδρομα μυελικής ανεπάρκειας Ιδιοπαθή (70%) – άγνωστης αιτιολογίας Ιδιοπαθή (70%) – άγνωστης αιτιολογίας Κληρονομούμενα (20%) Κληρονομούμενα (20%) Δευτεροπαθή (10%) Δευτεροπαθή (10%) Ακτινοβολία Ακτινοβολία Φάρμακα Φάρμακα Ιογενείς παράγοντες Ιογενείς παράγοντες Αυτοάνοσα νοσήματα (ΣΕΛ) Αυτοάνοσα νοσήματα (ΣΕΛ)
 – άγνωστης αιτιολογίας Ιδιοπαθή (70%) – άγνωστης αιτιολογίας Κληρονομούμενα (20%) Κληρονομούμενα (20%) Δευτεροπαθή (10%) Δευτεροπαθή (10%) Ακτινοβολία Ακτινοβολία Φάρμακα Φάρμακα Ιογενείς παράγοντες Ιογενείς παράγοντες Αυτοάνοσα νοσήματα (ΣΕΛ) Αυτοάνοσα νοσήματα (ΣΕΛ)")
4
Κληρονομούμενα σύνδρομα μυελικής ανεπάρκειας (ΚΣΜΑ) Θεωρούνται νοσήματα του παιδιατρικού πληθυσμού αν και σε μερικές περιπτώσεις η διάγνωση τίθεται όψιμα στην ενήλικη ζωή. Θεωρούνται νοσήματα του παιδιατρικού πληθυσμού αν και σε μερικές περιπτώσεις η διάγνωση τίθεται όψιμα στην ενήλικη ζωή. Αδυναμία προσδιορισμού της ακριβούς επίπτωσής τους Αδυναμία προσδιορισμού της ακριβούς επίπτωσής τους >25% των παιδιών και 10% των νεαρών ενηλίκων με επίκτητη απλαστική αναιμία πάσχουν από ΚΣΜΑ >25% των παιδιών και 10% των νεαρών ενηλίκων με επίκτητη απλαστική αναιμία πάσχουν από ΚΣΜΑ

5
Συχνότερα ΚΣΜΑ Αναιμία Fanconi Αναιμία Fanconi Συγγενής δυσκεράτωση Συγγενής δυσκεράτωση Σύνδρομο Shwachman – Diamond Σύνδρομο Shwachman – Diamond Αναιμία Blackfan – Diamond Αμεγακαρυοκυτταρική θρομβοπενία Αναιμία Blackfan – Diamond Αμεγακαρυοκυτταρική θρομβοπενία Σύνδρομο TAR (Thrombocytopenia-absent Radius) Σύνδρομο TAR (Thrombocytopenia-absent Radius) Συγγενής ουδετεροπενία Συγγενής ουδετεροπενία
 Σύνδρομο TAR (Thrombocytopenia-absent Radius) Συγγενής ουδετεροπενία Συγγενής ουδετεροπενία")
6
Διάγνωση ΚΣΜΑ Χαρακτηριστικά ευρήματα από την κλινική εξέταση Χαρακτηριστικά ευρήματα από την κλινική εξέταση Ασθενείς με: Ασθενείς με: «Επίκτητη» απλαστική αναιμία «Επίκτητη» απλαστική αναιμία Μυελοδυσπλαστικό Σ./ΟΜΛ Μυελοδυσπλαστικό Σ./ΟΜΛ Καρκίνο σε νεαρή ηλικία Καρκίνο σε νεαρή ηλικία Παρουσία χαρακτηριστικών μεταλλάξεων Παρουσία χαρακτηριστικών μεταλλάξεων

7
Ιστορία: Guido Fanconi Fanconi Anemia (Fanconi pancytopenia syndrome): 1927 – 3 αδέρφια με πανκυττταροπενία και ιδιαίτερα μορφολογικά χαρακτηριστικά Alter, FA101 (2006)
: 1927 – 3 αδέρφια με πανκυττταροπενία και ιδιαίτερα μορφολογικά χαρακτηριστικά Alter, FA101 (2006)")
8
Αναιμία Fanconi Κληρονομείται με: Κληρονομείται με: Αυτοσωματικό υπολειπόμενο χαρακτήρα Αυτοσωματικό υπολειπόμενο χαρακτήρα Φυλοσύνδετο υπολειπόμενο χαρακτήρα (σπάνια) Φυλοσύνδετο υπολειπόμενο χαρακτήρα (σπάνια) Χαρακτηρίζεται από χρωμοσωμική αστάθεια και αυξημένη προδιάθεση για κακοήθεια Χαρακτηρίζεται από χρωμοσωμική αστάθεια και αυξημένη προδιάθεση για κακοήθεια Διάγνωση συνήθως στην πρώτη δεκαετία ζωής Διάγνωση συνήθως στην πρώτη δεκαετία ζωής Επίπτωση 1 – 5/10 ⁶ πληθυσμού Επίπτωση 1 – 5/10 ⁶ πληθυσμού
 Φυλοσύνδετο υπολειπόμενο χαρακτήρα (σπάνια) Χαρακτηρίζεται από χρωμοσωμική αστάθεια και αυξημένη προδιάθεση για κακοήθεια Χαρακτηρίζεται από χρωμοσωμική αστάθεια και αυξημένη προδιάθεση για κακοήθεια Διάγνωση συνήθως στην πρώτη δεκαετία ζωής Διάγνωση συνήθως στην πρώτη δεκαετία ζωής Επίπτωση 1 – 5/10 ⁶ πληθυσμού Επίπτωση 1 – 5/10 ⁶ πληθυσμού")
9
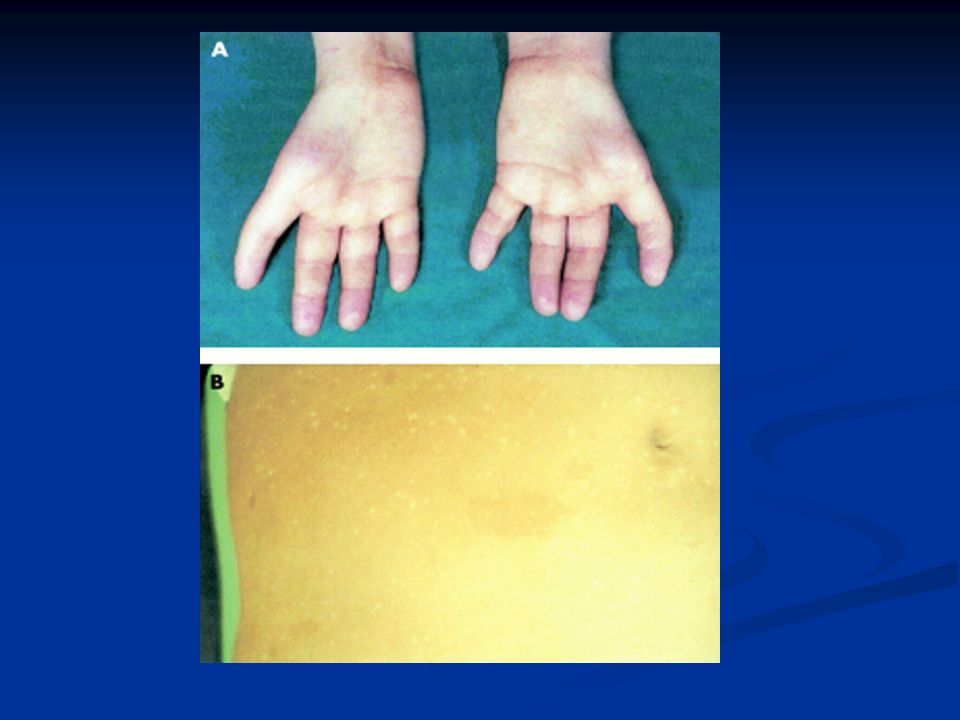
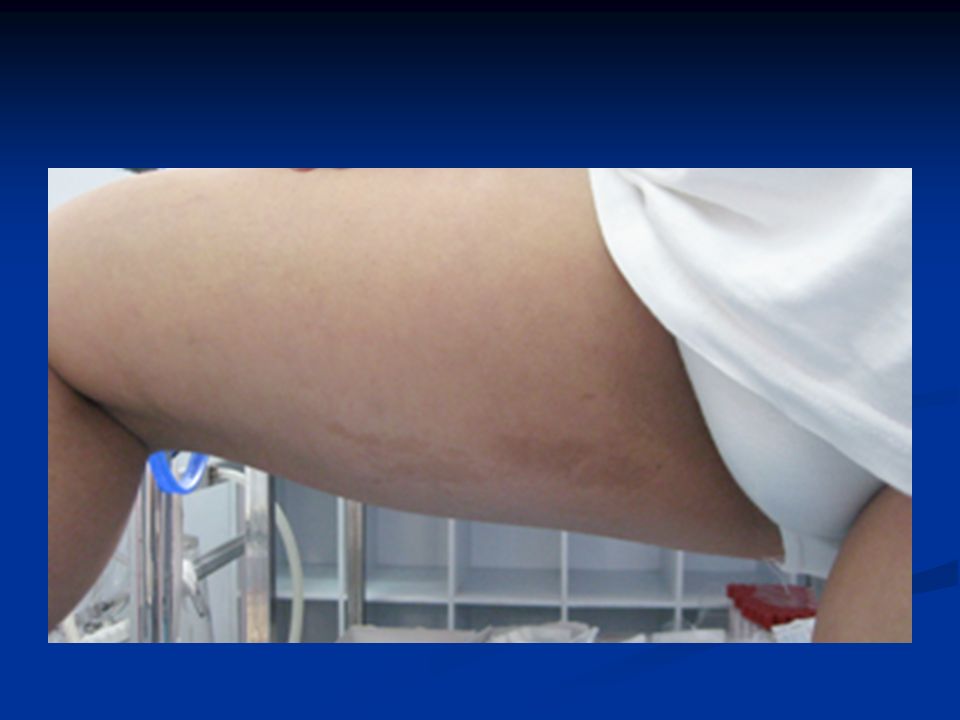
Κλινικά χαρακτηριστικά Κηλίδες café au lait Κηλίδες café au lait Πεταλοειδής νεφρός, ατρησία 12/λου Πεταλοειδής νεφρός, ατρησία 12/λου Καρδιακές /νευρολογικές ανωμαλίες Καρδιακές /νευρολογικές ανωμαλίες Κοντό ανάστημα Κοντό ανάστημα Μικροκεφαλία/μικροφθαλμία Μικροκεφαλία/μικροφθαλμία Ανώμαλοι αντίχειρες +/- υποπλασία κερκίδας Ανώμαλοι αντίχειρες +/- υποπλασία κερκίδας Υπογοναδισμός Υπογοναδισμός 25% χωρίς εμφανή χαρακτηριστικά 25% χωρίς εμφανή χαρακτηριστικά

10
Η μυελική ανεπάρκεια εμφανίζεται συνήθως την πρώτη δεκαετία ζωής Η μυελική ανεπάρκεια εμφανίζεται συνήθως την πρώτη δεκαετία ζωής Αίτια θανάντου: Αίτια θανάντου: Μυελική ανεπάρκεια Μυελική ανεπάρκεια ΟΜΛ / ΜΔΣ ΟΜΛ / ΜΔΣ Συμπαγών όγκων (επιθηλιακοί όγκοι κεφαλής, τραχήλου, γυναικολογικοί καρκίνοι, καρκίνος οισοφάγου, ήπατος, δέρματος κ. α. ) Συμπαγών όγκων (επιθηλιακοί όγκοι κεφαλής, τραχήλου, γυναικολογικοί καρκίνοι, καρκίνος οισοφάγου, ήπατος, δέρματος κ. α. ) 25% των ασθενών ο συμπαγής όγκος προηγείται της διάγνωσης της FA 25% των ασθενών ο συμπαγής όγκος προηγείται της διάγνωσης της FA
 Συμπαγών όγκων (επιθηλιακοί όγκοι κεφαλής, τραχήλου, γυναικολογικοί καρκίνοι, καρκίνος οισοφάγου, ήπατος, δέρματος κ. α. ) 25% των ασθενών ο συμπαγής όγκος προηγείται της διάγνωσης της FA 25% των ασθενών ο συμπαγής όγκος προηγείται της διάγνωσης της FA.")
11
Παθογένεια FA Ευθραυστότητα χρωμοσωμάτων Ευθραυστότητα χρωμοσωμάτων Ανώμαλη κινητική κυτταρικού κύκλου (παρατεταμένη G2 φάση) Ανώμαλη κινητική κυτταρικού κύκλου (παρατεταμένη G2 φάση) Αυξημένη απόπτωση Αυξημένη απόπτωση Βραχέα τελομερή Βραχέα τελομερή
 Ανώμαλη κινητική κυτταρικού κύκλου (παρατεταμένη G2 φάση) Αυξημένη απόπτωση Αυξημένη απόπτωση Βραχέα τελομερή Βραχέα τελομερή")
12
Γενετική ετερογένεια Έχουν περιγραφεί 15 ομάδες πρωτεϊνών (complementation groups) σε ασθενείς με FA με αντίστοιχα 15 χαρτογραφημένα γονίδια Έχουν περιγραφεί 15 ομάδες πρωτεϊνών (complementation groups) σε ασθενείς με FA με αντίστοιχα 15 χαρτογραφημένα γονίδια
 σε ασθενείς με FA με αντίστοιχα 15 χαρτογραφημένα γονίδια Έχουν περιγραφεί 15 ομάδες πρωτεϊνών (complementation groups) σε ασθενείς με FA με αντίστοιχα 15 χαρτογραφημένα γονίδια")
13
FA complementation groups/genetic subtypes Complementation group/gene Approximate % of FA patients Chromosome location Protein (amino acids) Exons A (FANCA)6516q24.31455 43 B (FANCBa)<1Xp22.2859 10 C (FANCC)129q22.3558 14 D1 (FANCD1b)<113q12.33418 27 D2 (FANCD2)<13p25.31451 44 E (FANCE)46p21.3536 10 F (FANCF)411p15374 1 G (FANCG)129p13622 14 I (FANCI)<115q26.11328 35 J (FANCJ/BRIP1c)<517q23.11249 20 L (FANCL)<12p16.1375 14 M (FANCM)<114q21.32048 23 N (FANCN/PALB2d)<116p12.1118613
 Exons A (FANCA)6516q B (FANCBa)<1Xp C (FANCC)129q D1 (FANCD1b)<113q D2 (FANCD2)<13p E (FANCE)46p F (FANCF)411p G (FANCG)129p I (FANCI)<115q J (FANCJ/BRIP1c)<517q L (FANCL)<12p M (FANCM)<114q N (FANCN/PALB2d)<116p")
14
84% των ασθενών υπάγονται στις κατηγορίες: Α, C, G 84% των ασθενών υπάγονται στις κατηγορίες: Α, C, G FANCD1 γονίδιο (BRCA2 tumor sup.) FANCD1 γονίδιο (BRCA2 tumor sup.) FANCJ (helicase/endonucleases) FANCJ (helicase/endonucleases) FANCM (DNA μετάθεση) FANCM (DNA μετάθεση)
 FANCD1 γονίδιο (BRCA2 tumor sup.) FANCJ (helicase/endonucleases) FANCJ (helicase/endonucleases) FANCM (DNA μετάθεση) FANCM (DNA μετάθεση)")
15
Δράση πρωτεϊνών FA

16
Διάγνωση Κλινική εξέταση Κλινική εξέταση Ατομικό ιστορικό Ατομικό ιστορικό Οικογενειακό ιστορικό Οικογενειακό ιστορικό Ειδικός εργαστηριακός έλεγχος Ειδικός εργαστηριακός έλεγχος

17
Διαγνωστικές δοκιμασίες Εξέταση περιφερικού αίματος Εξέταση περιφερικού αίματος Μυελόγραμμα, Οστεομυελική Βιοψία Μυελόγραμμα, Οστεομυελική Βιοψία Καρυότυπος μυελού Καρυότυπος μυελού Ειδικές διαγνωστικές εξετάσεις Ειδικές διαγνωστικές εξετάσεις

18
Οστεομυελική βιοψία, όπου ο λιπώδης ιστός επικρατεί (λευκά-διαυγή κύτταρα) ενώ υπάρχει ελάχιστη εκπροσώπιση του αιμοποιητικού ιαστού, ενδεικτικό μυελικής ανεπάρκειας
 ενώ υπάρχει ελάχιστη εκπροσώπιση του αιμοποιητικού ιαστού, ενδεικτικό μυελικής ανεπάρκειας")
19
Διαγνωστικές δοκιμασίες (Ι) Έλεγχος ευθραυστότητας χρωμοσωμάτων (DEB, MMC)(stress test) – Μωσαϊκισμός (ινοβλάστες) Έλεγχος ευθραυστότητας χρωμοσωμάτων (DEB, MMC)(stress test) – Μωσαϊκισμός (ινοβλάστες) Κυτταρομετρία ροής : προσδιορισμός G2 φάσης (παρατεταμένη) σε λεμφοκύτταρα Κυτταρομετρία ροής : προσδιορισμός G2 φάσης (παρατεταμένη) σε λεμφοκύτταρα
 Έλεγχος ευθραυστότητας χρωμοσωμάτων (DEB, MMC)(stress test) – Μωσαϊκισμός (ινοβλάστες) Έλεγχος ευθραυστότητας χρωμοσωμάτων (DEB, MMC)(stress test) – Μωσαϊκισμός (ινοβλάστες) Κυτταρομετρία ροής : προσδιορισμός G2 φάσης (παρατεταμένη) σε λεμφοκύτταρα Κυτταρομετρία ροής : προσδιορισμός G2 φάσης (παρατεταμένη) σε λεμφοκύτταρα")
20
Chromosome breakage in Fanconi Anemia cells FA cells were treated with mitomycin C and harvested in metaphase. Typical abnormalities include radial formation (green circle) and chromosome breaks (red arrows).
 and chromosome breaks (red arrows)..")
21
Διαγνωστικές δοκιμασίες (ΙΙ) Ανίχνευση FANCD2-L με Western blot ανάλυση: Ανίχνευση FANCD2-L με Western blot ανάλυση: Απουσία ubiquitinated FANCD2 → διαταραχή στη συγκρότηση του πρωτεϊνικού πυρήνα Απουσία ubiquitinated FANCD2 → διαταραχή στη συγκρότηση του πρωτεϊνικού πυρήνα Παρουσία ubiquitinated FANCD2 → μεταλλάξεις στα γονίδια FANCD1 ή FANCJ Παρουσία ubiquitinated FANCD2 → μεταλλάξεις στα γονίδια FANCD1 ή FANCJ Ανίχνευση των complementation groups: χρήση λεμφοκύτταρα περιφ. αίματος FA Ανίχνευση των complementation groups: χρήση λεμφοκύτταρα περιφ. αίματος FA Οριστική διάγνωση: αναζήτηση των γνωστών γονιδίων Οριστική διάγνωση: αναζήτηση των γνωστών γονιδίων

22
Σχέση γονότυπου/φαινότυπου στην FA FA – C : πτωχή πρόγνωση FA – C : πτωχή πρόγνωση FACC mutation : πολλαπλές γενετικές ανωμαλίες και πρώιμες αιματολογικές εκδηλώσεις στους εβραίους Εσκενάζυ σε αντίθεση με την ίδια μετάλλαξη στους Ιάπωνες FACC mutation : πολλαπλές γενετικές ανωμαλίες και πρώιμες αιματολογικές εκδηλώσεις στους εβραίους Εσκενάζυ σε αντίθεση με την ίδια μετάλλαξη στους Ιάπωνες FA – D1, N: (ομόζυγη μετάλλαξη στο BRCA2) πρώιμη ανάπτυξη κακοηθειών (μυελοβλάστωμα, όγκος Wilms, ΟΜΛ). FA – D1, N: (ομόζυγη μετάλλαξη στο BRCA2) πρώιμη ανάπτυξη κακοηθειών (μυελοβλάστωμα, όγκος Wilms, ΟΜΛ).
 πρώιμη ανάπτυξη κακοηθειών (μυελοβλάστωμα, όγκος Wilms, ΟΜΛ)..")
23
Σχέση γονότυπου/φαινότυπου FA-D1 (BRCA2) FA-D1 (BRCA2) FA-N καρκίνος μαστού FA-N καρκίνος μαστού FA-J FA-J
 FA-D1 (BRCA2) FA-N καρκίνος μαστού FA-N καρκίνος μαστού FA-J FA-J")
24
Θεραπεία FA Κορτικοστεροειδή Κορτικοστεροειδή Ανδρογόνα (οξυμεθολόνη) Ανδρογόνα (οξυμεθολόνη) Αλλογενής ΜΜΟ Αλλογενής ΜΜΟ Γονιδιακή θεραπεία (?) Γονιδιακή θεραπεία (?)
 Ανδρογόνα (οξυμεθολόνη) Αλλογενής ΜΜΟ Αλλογενής ΜΜΟ Γονιδιακή θεραπεία ( ) Γονιδιακή θεραπεία ( )")
25
Οξυμεθολόνη Απάντηση του 50-70% των αθενών Απάντηση του 50-70% των αθενών Ανάπτυξη αντοχής Ανάπτυξη αντοχής Αρρενοποίηση θηλέων Αρρενοποίηση θηλέων Ηπατοτοξικότητα Ηπατοτοξικότητα

26
Μεταμόσχευση SC Οριστική θεραπεία της μυελικής ανεπάρκειας Οριστική θεραπεία της μυελικής ανεπάρκειας Διετής επιβίωση: Διετής επιβίωση: Συγγενής συμβατός δότης: 70% Συγγενής συμβατός δότης: 70% Μη συγγενής συμβατός δότης: 20-40% Μη συγγενής συμβατός δότης: 20-40% Τροποποιημένο πρωτόκολλο Τροποποιημένο πρωτόκολλο δόση κυκλοφωσφαμίδης και ακτινοβολίας δόση κυκλοφωσφαμίδης και ακτινοβολίας Χρήση fludarabine Χρήση fludarabine Διετής επιβίωση 65-90% Διετής επιβίωση 65-90%

27
Γονιδιακή θεραπεία Μελλοντικός στόχος Μελλοντικός στόχος Παροχή πολ/στικού πλεονεκτήματος των τροποποιημένων κυττάρων έναντι του λοιπού υποκυτταρικού πληθυσμού του μυελού Παροχή πολ/στικού πλεονεκτήματος των τροποποιημένων κυττάρων έναντι του λοιπού υποκυτταρικού πληθυσμού του μυελού

33
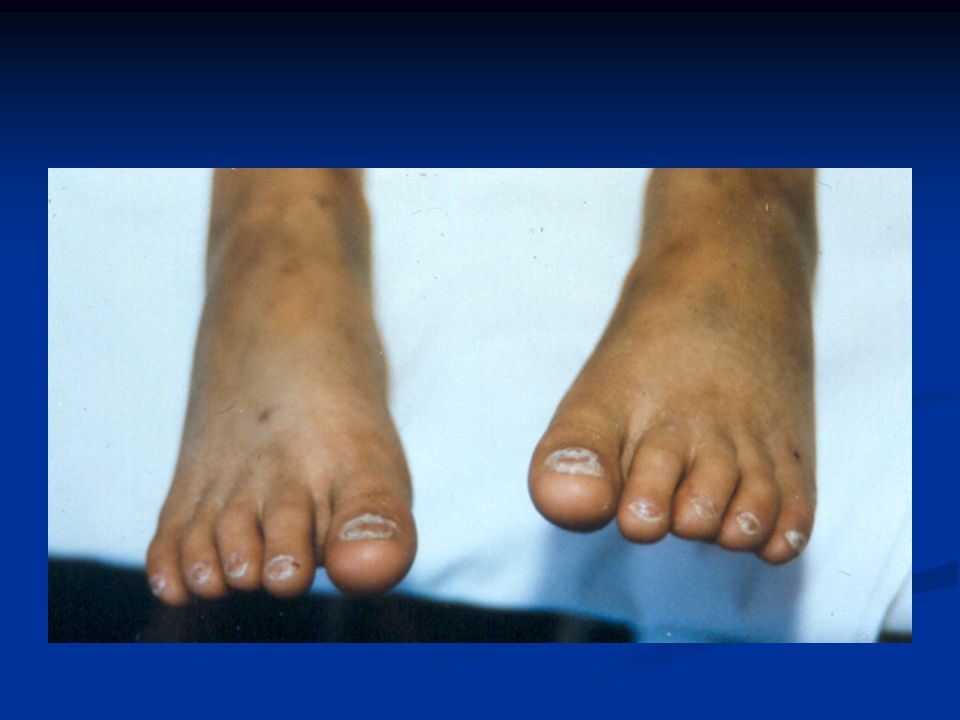
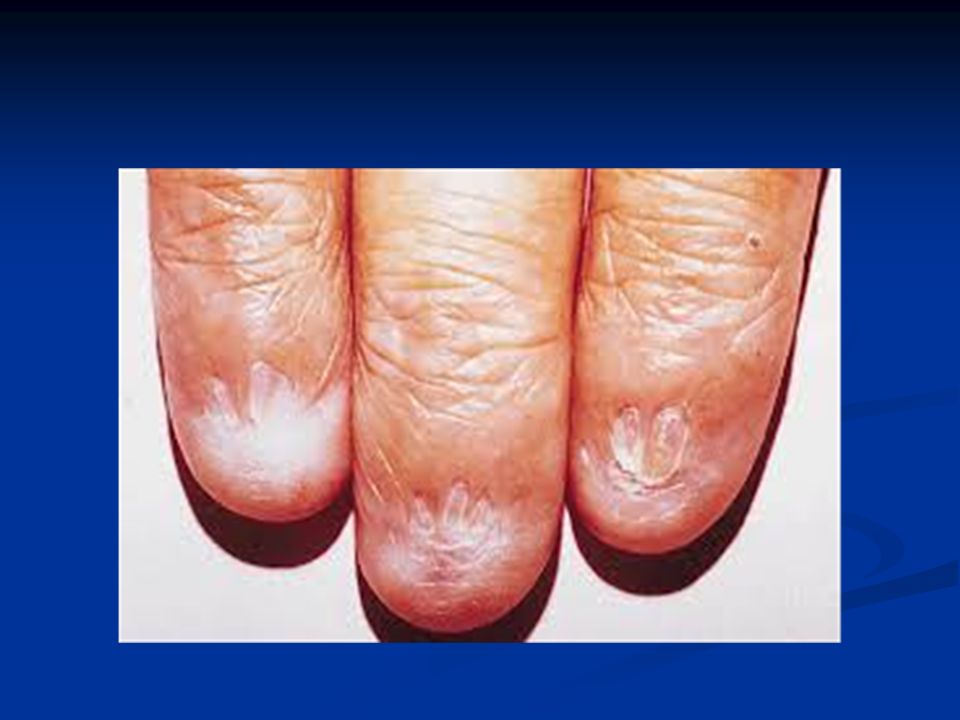
Συγγενής Δυσκεράτωση Κληρονομικό Σ. Μυελικής Ανεπάρκειας που χαρακτηρίζεται από συγκεκριμένες βλεννογονοδερματικές αλλοιώσεις: Κληρονομικό Σ. Μυελικής Ανεπάρκειας που χαρακτηρίζεται από συγκεκριμένες βλεννογονοδερματικές αλλοιώσεις: Διαταραχές χρωστικής δέρματος Διαταραχές χρωστικής δέρματος Ονυχοδυστροφία Ονυχοδυστροφία Λευκοπλακία στομ. βλεννογόνου Λευκοπλακία στομ. βλεννογόνου Επίπτωση: 1/10 6 ατόμων Επίπτωση: 1/10 6 ατόμων Άλλες συνήθεις ανωμαλίες (δόντια, γαστρ/κο, ουροποιητικό, νευρικό, οφθαλμοί, αναπνευστικό και σκελετικό) Άλλες συνήθεις ανωμαλίες (δόντια, γαστρ/κο, ουροποιητικό, νευρικό, οφθαλμοί, αναπνευστικό και σκελετικό)
 Άλλες συνήθεις ανωμαλίες (δόντια, γαστρ/κο, ουροποιητικό, νευρικό, οφθαλμοί, αναπνευστικό και σκελετικό).")
34
Κληρονομείται με: Κληρονομείται με: Φυλοσύνδετο υπολειπόμενο χαρακτήρα (Xq28) Φυλοσύνδετο υπολειπόμενο χαρακτήρα (Xq28) Αυτοσωματικό επικρατητικό χαρακτήρα Αυτοσωματικό επικρατητικό χαρακτήρα Αυτοσωματικό υπολειπόμενο χαρακτήρα Αυτοσωματικό υπολειπόμενο χαρακτήρα Σποραδικές μεταλλάξεις Σποραδικές μεταλλάξεις Άιτια θανάτου: Άιτια θανάτου: Μυελική ανεπάρκεια (70% - τρίτη δεκαετία) Μυελική ανεπάρκεια (70% - τρίτη δεκαετία) Ανάπτυξη κακοηθειών (10 - 15%) Ανάπτυξη κακοηθειών (10 - 15%) Πνευμονική προσβολή (10%) Πνευμονική προσβολή (10%)
 Φυλοσύνδετο υπολειπόμενο χαρακτήρα (Xq28) Αυτοσωματικό επικρατητικό χαρακτήρα Αυτοσωματικό επικρατητικό χαρακτήρα Αυτοσωματικό υπολειπόμενο χαρακτήρα Αυτοσωματικό υπολειπόμενο χαρακτήρα Σποραδικές μεταλλάξεις Σποραδικές μεταλλάξεις Άιτια θανάτου: Άιτια θανάτου: Μυελική ανεπάρκεια (70% - τρίτη δεκαετία) Μυελική ανεπάρκεια (70% - τρίτη δεκαετία) Ανάπτυξη κακοηθειών ( %) Ανάπτυξη κακοηθειών ( %) Πνευμονική προσβολή (10%) Πνευμονική προσβολή (10%)")
35
Κλινικές εκδηλώσεις Πρώτη δεκαετία : διαταραχές ονύχων – δέρματος Πρώτη δεκαετία : διαταραχές ονύχων – δέρματος Δεύτερη δεκαετία: μυελική ανεπάρκεια (90% των ασθενών θα αναπτύξουν μυελική ανεπάρκεια έως την ηλικία των 30 ετών) Δεύτερη δεκαετία: μυελική ανεπάρκεια (90% των ασθενών θα αναπτύξουν μυελική ανεπάρκεια έως την ηλικία των 30 ετών) Σπανίως η μυελική ανεπάρκεια προηγείται των δερματικών εκδηλώσεων Σπανίως η μυελική ανεπάρκεια προηγείται των δερματικών εκδηλώσεων
 Δεύτερη δεκαετία: μυελική ανεπάρκεια (90% των ασθενών θα αναπτύξουν μυελική ανεπάρκεια έως την ηλικία των 30 ετών) Σπανίως η μυελική ανεπάρκεια προηγείται των δερματικών εκδηλώσεων Σπανίως η μυελική ανεπάρκεια προηγείται των δερματικών εκδηλώσεων")
36
Κλινική εικόνα Ατρησία δακρυικών πόρων 30% Ατρησία δακρυικών πόρων 30% Μαθησιακές δυσκολίες, αναπτυξιακή / νοητική καθυστέρηση 25.4% Μαθησιακές δυσκολίες, αναπτυξιακή / νοητική καθυστέρηση 25.4% Πνευμονική νόσος 20.3% Πνευμονική νόσος 20.3% Κοντό ανάστημα 19.5% Κοντό ανάστημα 19.5% Εκτεταμένη οδοντική καταστροφή 16.9% Εκτεταμένη οδοντική καταστροφή 16.9% Οισοφαγική στένωση 16.9% Οισοφαγική στένωση 16.9% Πρόωρη απώλεια μαλλιών, γκριζάρισμα,αραιές βλεφαρίδες 16.1% Υπεριδρωσία 15.3% Κακοήθεια 9.8% Ενδομήτρια καθυστέρηση ανάπτυξης 7.6% Ηπατοπάθεια, εντεροπάθεια, πεπτικό έλκος 7.3%

40
Παθογένεια Πρόκειται για σύνδρομο χαρακτηριζόμενο από βραχύ μήκος τελομερών Πρόκειται για σύνδρομο χαρακτηριζόμενο από βραχύ μήκος τελομερών Μελέτες των προγονικών αιμοποιητικών κυττάρων ανέδειξαν μειωμένο αριθμό αυτών ακόμα και στις περιπτώσεις όπου οι μετρήσεις του ΠΑ ήταν εντός φυσιολογικών ορίων → Βλάβη στο αρχέγονο στελεχοκύτταρο Μελέτες των προγονικών αιμοποιητικών κυττάρων ανέδειξαν μειωμένο αριθμό αυτών ακόμα και στις περιπτώσεις όπου οι μετρήσεις του ΠΑ ήταν εντός φυσιολογικών ορίων → Βλάβη στο αρχέγονο στελεχοκύτταρο

41
Τελομερή Χαρακτηριστικές επαναλήψεις αμινοξέων (TTAGGG) εντοπιζόμενες στα άκρα των χρωμοσωμάτων Χαρακτηριστικές επαναλήψεις αμινοξέων (TTAGGG) εντοπιζόμενες στα άκρα των χρωμοσωμάτων Προστατεύουν την ακεραιότητα των χρωμοσωμάτων κατά τη φάση της αντιγραφής Προστατεύουν την ακεραιότητα των χρωμοσωμάτων κατά τη φάση της αντιγραφής Το μήκος των τελομερών διατηρείται σταθερό από τη δράση της τελομεράσης Το μήκος των τελομερών διατηρείται σταθερό από τη δράση της τελομεράσης
 εντοπιζόμενες στα άκρα των χρωμοσωμάτων Χαρακτηριστικές επαναλήψεις αμινοξέων (TTAGGG) εντοπιζόμενες στα άκρα των χρωμοσωμάτων Προστατεύουν την ακεραιότητα των χρωμοσωμάτων κατά τη φάση της αντιγραφής Προστατεύουν την ακεραιότητα των χρωμοσωμάτων κατά τη φάση της αντιγραφής Το μήκος των τελομερών διατηρείται σταθερό από τη δράση της τελομεράσης Το μήκος των τελομερών διατηρείται σταθερό από τη δράση της τελομεράσης")
42
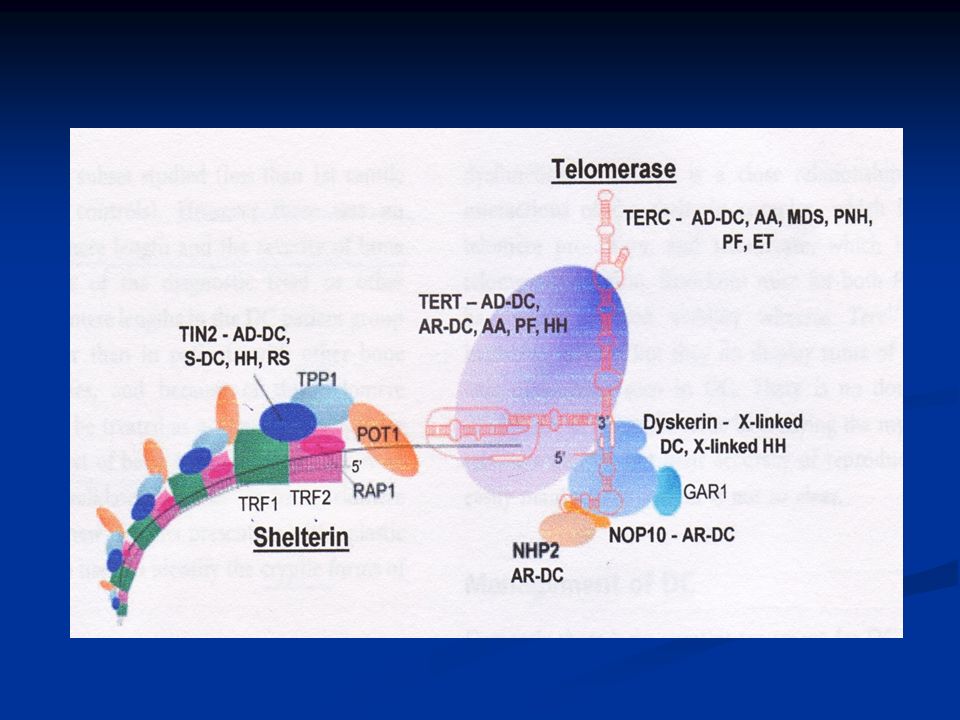
Τελομεράση Τελομεράση: ενζυμικό σύμπλεγμα ριβονουκλεοπρωτεϊνών, υπεύθυνο για τη διατήρηση του μήκους των τελομερών. Τελομεράση: ενζυμικό σύμπλεγμα ριβονουκλεοπρωτεϊνών, υπεύθυνο για τη διατήρηση του μήκους των τελομερών. ↑ έκφραση σε ιστούς με έντονη αναγεννητική δραστηριότητα ↑ έκφραση σε ιστούς με έντονη αναγεννητική δραστηριότητα Αποτελείται από 5 συστατικά: Αποτελείται από 5 συστατικά: TERC, TERT, Dyskerin,NOP10, NHP2 TERC, TERT, Dyskerin,NOP10, NHP2

43
Φυλοσύνδετος υπολειπόμενος τύπος Η πιο σοβαρή μορφή με πρώιμες κλινικές εκδηλώσεις Η πιο σοβαρή μορφή με πρώιμες κλινικές εκδηλώσεις Υπεύθυνο γονίδιο DKC1 (Xq28)→Dyskerin Υπεύθυνο γονίδιο DKC1 (Xq28)→Dyskerin Εκφράζεται σε όλους τους ιστούς και δικαιολογεί την κλινική εικόνα της DC Εκφράζεται σε όλους τους ιστούς και δικαιολογεί την κλινική εικόνα της DC
→Dyskerin Υπεύθυνο γονίδιο DKC1 (Xq28)→Dyskerin Εκφράζεται σε όλους τους ιστούς και δικαιολογεί την κλινική εικόνα της DC Εκφράζεται σε όλους τους ιστούς και δικαιολογεί την κλινική εικόνα της DC")
44
Δράσεις Dyskerin Μαζί με τα snoRNAs ρυθμίζει τη μετατροπή ουρακίλης→ψευδοουρακίλη του ριβοσωμικού RNA (rRNA). Διαδικασία σημαντική για τη σύνθεση των ριβοσωμάτων Μαζί με τα snoRNAs ρυθμίζει τη μετατροπή ουρακίλης→ψευδοουρακίλη του ριβοσωμικού RNA (rRNA). Διαδικασία σημαντική για τη σύνθεση των ριβοσωμάτων Συνεργάζεται με το RNA συστατικό της τελομεράσης(TERC) Συνεργάζεται με το RNA συστατικό της τελομεράσης(TERC)
. Διαδικασία σημαντική για τη σύνθεση των ριβοσωμάτων Συνεργάζεται με το RNA συστατικό της τελομεράσης(TERC) Συνεργάζεται με το RNA συστατικό της τελομεράσης(TERC).")
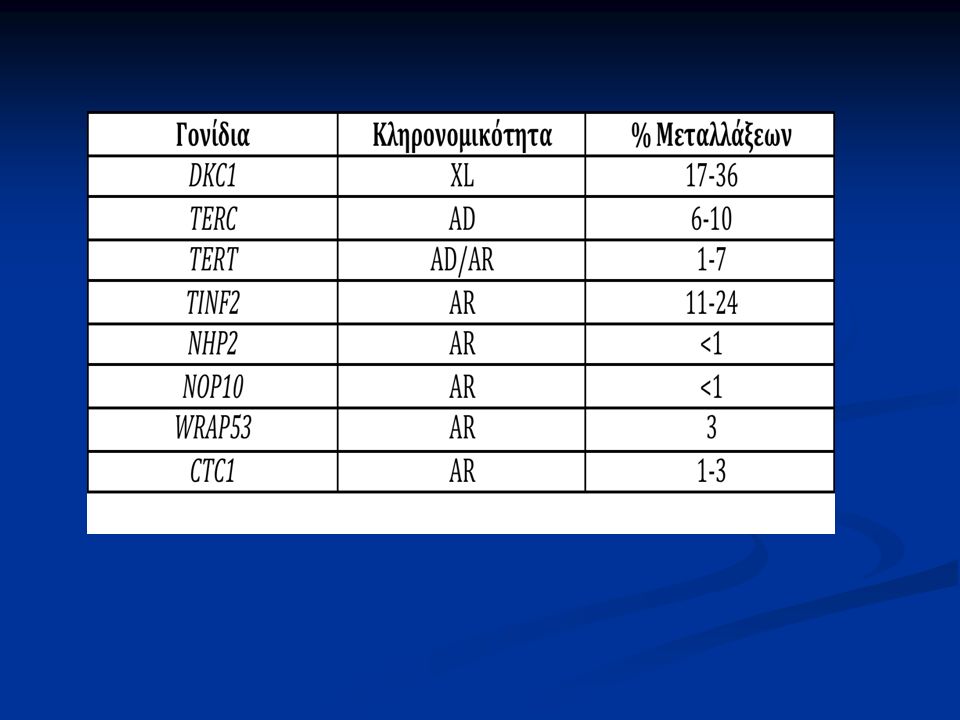
45
Αυτοσωματικός επικρατητικός χαρακτήρας Μεταλλάξεις του: Μεταλλάξεις του: TERC γονιδίου (3q26)→πρωτεΐνη TERC TERC γονιδίου (3q26)→πρωτεΐνη TERC TERT γονιδίου (5p15) → πρωτεΐνη TERT TERT γονιδίου (5p15) → πρωτεΐνη TERT Εκδηλώσεις από τους βλεννογόνους μπορεί να απουσιάζουν ή να είναι ήπιες Εκδηλώσεις από τους βλεννογόνους μπορεί να απουσιάζουν ή να είναι ήπιες
→πρωτεΐνη TERC TERC γονιδίου (3q26)→πρωτεΐνη TERC TERT γονιδίου (5p15) → πρωτεΐνη TERT TERT γονιδίου (5p15) → πρωτεΐνη TERT Εκδηλώσεις από τους βλεννογόνους μπορεί να απουσιάζουν ή να είναι ήπιες Εκδηλώσεις από τους βλεννογόνους μπορεί να απουσιάζουν ή να είναι ήπιες")
46
Αυτοσωματικός υπολειπόμενος χαρακτήρας Μεταλλάξεις των πρωτεϊνών του συμπλέγματος H/ACA: Μεταλλάξεις των πρωτεϊνών του συμπλέγματος H/ACA: NOP10 γονιδίου (15q14)→πρωτεΐνη NOP10 NOP10 γονιδίου (15q14)→πρωτεΐνη NOP10 NHP2 γονίδιο → πρωτεΐνη NHP2 NHP2 γονίδιο → πρωτεΐνη NHP2 TERT γονιδίου (5p15) → πρωτεΐνη TERT TERT γονιδίου (5p15) → πρωτεΐνη TERT Ομόζυγη μετάλλαξη στο TERT → Hoyeraal- Hreidarsson σύνδρομο (διαταραχές αύξησης, νευρολογικές διαταραχές, μυελική ανεπάρκεια, ανοσοανεπάρκεια) Ομόζυγη μετάλλαξη στο TERT → Hoyeraal- Hreidarsson σύνδρομο (διαταραχές αύξησης, νευρολογικές διαταραχές, μυελική ανεπάρκεια, ανοσοανεπάρκεια)
→πρωτεΐνη NOP10 NOP10 γονιδίου (15q14)→πρωτεΐνη NOP10 NHP2 γονίδιο → πρωτεΐνη NHP2 NHP2 γονίδιο → πρωτεΐνη NHP2 TERT γονιδίου (5p15) → πρωτεΐνη TERT TERT γονιδίου (5p15) → πρωτεΐνη TERT Ομόζυγη μετάλλαξη στο TERT → Hoyeraal- Hreidarsson σύνδρομο (διαταραχές αύξησης, νευρολογικές διαταραχές, μυελική ανεπάρκεια, ανοσοανεπάρκεια) Ομόζυγη μετάλλαξη στο TERT → Hoyeraal- Hreidarsson σύνδρομο (διαταραχές αύξησης, νευρολογικές διαταραχές, μυελική ανεπάρκεια, ανοσοανεπάρκεια)")
47
Σποραδικές περιπτώσεις Ο ρόλος της Shelterin Σύμπλεγμα 6 πρωτεϊνών 1. Καθορισμός της δομής του άκρου των τελομερών 2. Συμβολή στη δημιουργία των t – loops 3. Ρύθμιση της σύνθεσης του DNA των τελομερών από την τελομεράση

48
Shelterin και DC Μετάλλαξη σε μία πρωτεΐνη του συμπλέγματος της Shelterin (TINF2) έχει βρεθεί σε ασθενείς με DC. Μετάλλαξη σε μία πρωτεΐνη του συμπλέγματος της Shelterin (TINF2) έχει βρεθεί σε ασθενείς με DC. Η συγκεκριμένη μετάλλαξη οδηγεί στη δημιουργία εξαιρετικά βραχέων τελομερών και συνοδεύεται από βαριά κλινική εικόνα Η συγκεκριμένη μετάλλαξη οδηγεί στη δημιουργία εξαιρετικά βραχέων τελομερών και συνοδεύεται από βαριά κλινική εικόνα Πρόκειται για de novo μετάλλαξη Πρόκειται για de novo μετάλλαξη
 έχει βρεθεί σε ασθενείς με DC. Η συγκεκριμένη μετάλλαξη οδηγεί στη δημιουργία εξαιρετικά βραχέων τελομερών και συνοδεύεται από βαριά κλινική εικόνα Η συγκεκριμένη μετάλλαξη οδηγεί στη δημιουργία εξαιρετικά βραχέων τελομερών και συνοδεύεται από βαριά κλινική εικόνα Πρόκειται για de novo μετάλλαξη Πρόκειται για de novo μετάλλαξη.")
52
Διαγνωστικές δοκιμασίες Προσδιορισμός μήκους τελομερών (FISH- flow cytometry) Προσδιορισμός μήκους τελομερών (FISH- flow cytometry) Αναζήτηση των σχετιζόμενων με τη νόσο γονιδίων Αναζήτηση των σχετιζόμενων με τη νόσο γονιδίων
 Προσδιορισμός μήκους τελομερών (FISH- flow cytometry) Αναζήτηση των σχετιζόμενων με τη νόσο γονιδίων Αναζήτηση των σχετιζόμενων με τη νόσο γονιδίων")
53
Θεραπεία Οξυμεθολόνη Οξυμεθολόνη G-CSF, EPO G-CSF, EPO Αλλογενής ΜΜΟ Αλλογενής ΜΜΟ σοβαρές επιπλοκές από το αναπνευστικό, αποφυγή χρήσης busulphan και ακτινοβολίας σοβαρές επιπλοκές από το αναπνευστικό, αποφυγή χρήσης busulphan και ακτινοβολίας Ιδανικός υποψήφιος: χωρίς πνευμονική προσβολή, διαθέσιμος συμβ. συγγενής δότης Ιδανικός υποψήφιος: χωρίς πνευμονική προσβολή, διαθέσιμος συμβ. συγγενής δότης Γονιδιακή θεραπεία (μελλοντικός στόχος) Γονιδιακή θεραπεία (μελλοντικός στόχος)
 Γονιδιακή θεραπεία (μελλοντικός στόχος).")
54
Σ.Shwachman – Diamond Περιγράφηκε το 1964. Περιγράφηκε το 1964. Κληρονομείται με αυτοσωματικό υπολειπόμενο χαρακτήρα Κληρονομείται με αυτοσωματικό υπολειπόμενο χαρακτήρα Χαρακτηρίζεται από: Χαρακτηρίζεται από: Ανεπάρκεια εξωκρινούς μοίρας παγκρέατος Ανεπάρκεια εξωκρινούς μοίρας παγκρέατος Μυελική ανεπάρκεια (20% πανκυτταροπενία), ΜΔΣ, ΟΜΛ (25%, M:F=3:1) Μυελική ανεπάρκεια (20% πανκυτταροπενία), ΜΔΣ, ΟΜΛ (25%, M:F=3:1) Σωματικές ανωμαλίες (μεταφυσιακή χονδροδυσπλασία -75%) Σωματικές ανωμαλίες (μεταφυσιακή χονδροδυσπλασία -75%)
, ΜΔΣ, ΟΜΛ (25%, M:F=3:1) Μυελική ανεπάρκεια (20% πανκυτταροπενία), ΜΔΣ, ΟΜΛ (25%, M:F=3:1) Σωματικές ανωμαλίες (μεταφυσιακή χονδροδυσπλασία -75%) Σωματικές ανωμαλίες (μεταφυσιακή χονδροδυσπλασία -75%).")
56
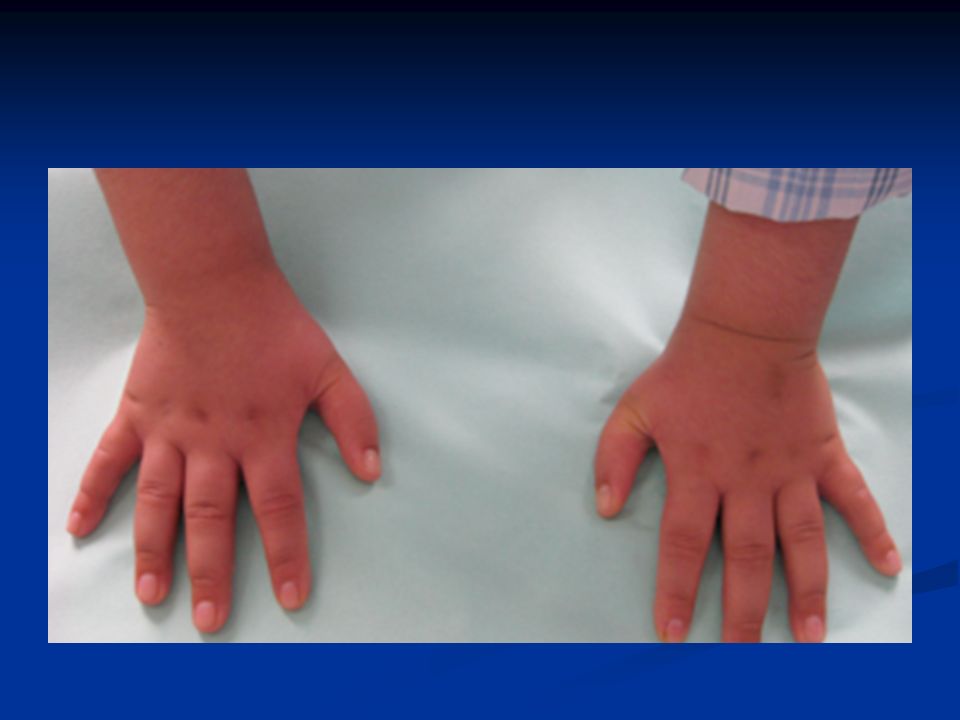
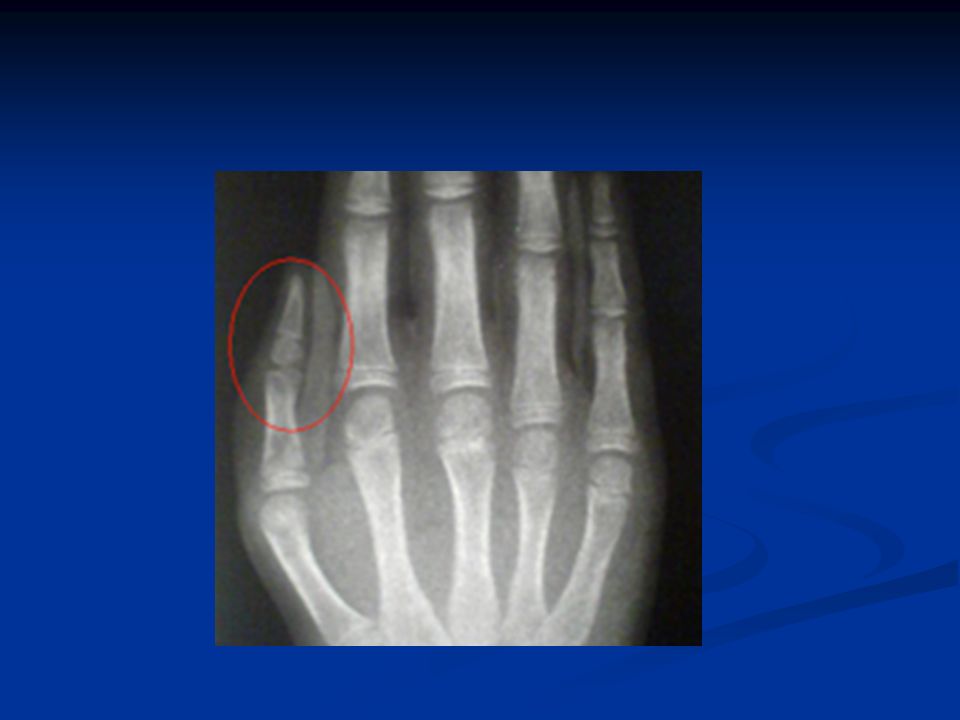
Κλινική εικόνα Ανεπαρκής αύξηση (δυσαπορρόφηση) Ανεπαρκής αύξηση (δυσαπορρόφηση) Κοντό ανάστημα Κοντό ανάστημα Ηπατομεγαλία Ηπατομεγαλία Ανωμαλίες θωρακικού κλωβού Ανωμαλίες θωρακικού κλωβού Υπερτελωρισμός Υπερτελωρισμός Συνδακτυλία Συνδακτυλία Θολωτή υπερώα Θολωτή υπερώα Δυσπλασία οδόντων Δυσπλασία οδόντων
 Ανεπαρκής αύξηση (δυσαπορρόφηση) Κοντό ανάστημα Κοντό ανάστημα Ηπατομεγαλία Ηπατομεγαλία Ανωμαλίες θωρακικού κλωβού Ανωμαλίες θωρακικού κλωβού Υπερτελωρισμός Υπερτελωρισμός Συνδακτυλία Συνδακτυλία Θολωτή υπερώα Θολωτή υπερώα Δυσπλασία οδόντων Δυσπλασία οδόντων")
57
2003: γονίδιο SBDS (7q11) 2003: γονίδιο SBDS (7q11) >90% των ασθενών φέρουν μεταλλάξεις στο SBDS >90% των ασθενών φέρουν μεταλλάξεις στο SBDS 10% άλλο γονίδιο – άγνωστο έως σήμερα 10% άλλο γονίδιο – άγνωστο έως σήμερα
 2003: γονίδιο SBDS (7q11) >90% των ασθενών φέρουν μεταλλάξεις στο SBDS >90% των ασθενών φέρουν μεταλλάξεις στο SBDS 10% άλλο γονίδιο – άγνωστο έως σήμερα 10% άλλο γονίδιο – άγνωστο έως σήμερα")
58
Λειτουργία SBDS γονιδίου (7q11) Παραγωγή πρωτεΐνης η οποία εμπλέκεται στην ωρίμανση της 60s ριβοσωμικής υπομονάδας Παραγωγή πρωτεΐνης η οποία εμπλέκεται στην ωρίμανση της 60s ριβοσωμικής υπομονάδας Αυξημένη απόπτωση και βραχέα τελομερή Αυξημένη απόπτωση και βραχέα τελομερή↓ Δυσλειτουργία ριβοσωμάτων
 Παραγωγή πρωτεΐνης η οποία εμπλέκεται στην ωρίμανση της 60s ριβοσωμικής υπομονάδας Παραγωγή πρωτεΐνης η οποία εμπλέκεται στην ωρίμανση της 60s ριβοσωμικής υπομονάδας Αυξημένη απόπτωση και βραχέα τελομερή Αυξημένη απόπτωση και βραχέα τελομερή↓ Δυσλειτουργία ριβοσωμάτων")
59
Διαγνωστικές δοκιμασίες Ουδετεροπενία επιμένουσα Ουδετεροπενία επιμένουσα 20% πανκυτταροπενία 20% πανκυτταροπενία Εξωκρινής παγκρεατική λειτουργία Εξωκρινής παγκρεατική λειτουργία Θρυψινογόνο ορού Θρυψινογόνο ορού Ισοαμυλάση ορού Ισοαμυλάση ορού Ανίχνευση του SBDS γονιδίου Ανίχνευση του SBDS γονιδίου

60
Θεραπεία Δυσαπορρόφηση → χορήγηση παγκρεατικών ενζύμων (ύφεση 50% των ασθενών μετά την ηλικία των 5 ετών) Δυσαπορρόφηση → χορήγηση παγκρεατικών ενζύμων (ύφεση 50% των ασθενών μετά την ηλικία των 5 ετών) Ουδετεροπενία → G-CSF Ουδετεροπενία → G-CSF Οξυμεθολόνη Οξυμεθολόνη Αλλογενής ΜΜΟ ( 25% ΟΜΛ, Α:Γ=3:1) Αλλογενής ΜΜΟ ( 25% ΟΜΛ, Α:Γ=3:1) Μη αιματολογικές κακοήθειες δεν έχουν αναφερθεί στους ασθενείς με σ. SD Μη αιματολογικές κακοήθειες δεν έχουν αναφερθεί στους ασθενείς με σ. SD

61
Αναιμία Diamond - Blackfan Περιγράφηκε για πρώτη φορά το 1938 (συγγενής απλασία της ερυθράς σειράς) Περιγράφηκε για πρώτη φορά το 1938 (συγγενής απλασία της ερυθράς σειράς) Ετήσια επίπτωση: ≈ 5/10 ⁶ γεννήσεις ζώντων Ετήσια επίπτωση: ≈ 5/10 ⁶ γεννήσεις ζώντων Εκδηλώνεται κατά την πρώιμη βρεφική ηλικία με χαρακτηριστική εκλεκτική μείωση των πρόδρομων της ερυθράς σειράς και ορθόχρωμη μακροκυτταρική αναιμία Εκδηλώνεται κατά την πρώιμη βρεφική ηλικία με χαρακτηριστική εκλεκτική μείωση των πρόδρομων της ερυθράς σειράς και ορθόχρωμη μακροκυτταρική αναιμία
 Περιγράφηκε για πρώτη φορά το 1938 (συγγενής απλασία της ερυθράς σειράς) Ετήσια επίπτωση: ≈ 5/10 ⁶ γεννήσεις ζώντων Ετήσια επίπτωση: ≈ 5/10 ⁶ γεννήσεις ζώντων Εκδηλώνεται κατά την πρώιμη βρεφική ηλικία με χαρακτηριστική εκλεκτική μείωση των πρόδρομων της ερυθράς σειράς και ορθόχρωμη μακροκυτταρική αναιμία Εκδηλώνεται κατά την πρώιμη βρεφική ηλικία με χαρακτηριστική εκλεκτική μείωση των πρόδρομων της ερυθράς σειράς και ορθόχρωμη μακροκυτταρική αναιμία")
62
Στοιχεία κλινικής εξέτασης Κρανιοπροσωπικές δυσπλασίες Κρανιοπροσωπικές δυσπλασίες Δυσπλασίες αντίχειρα Δυσπλασίες αντίχειρα Καρδιακές ανωμαλίες Καρδιακές ανωμαλίες Ανωμαλίες του ουρογεννητικού συστήματος Ανωμαλίες του ουρογεννητικού συστήματος Χαμηλό ανάστημα Χαμηλό ανάστημα

64
Κριτήρια διάγνωσης 1. Ορθόχρωμη, μακροκυτταρική αναιμία 2. ↓ ΔΕΚ 3. Φυσιολογική κυτταροβρίθεια μυελού με μείωση μόνο της ερυθράς σειράς (ΕΒ<5%) 4. Φυσιολογικός ή ↗ αριθμός λευκοκυττάρων 5. Φυσιολογικός ή ↗ αριθμός αιμοπεταλίων 6. ↑ eADA, ↑Hb F
 4. Φυσιολογικός ή ↗ αριθμός λευκοκυττάρων 5. Φυσιολογικός ή ↗ αριθμός αιμοπεταλίων 6. ↑ eADA, ↑Hb F.")
65
Κληρονομικότητα Αυτοσωματικός επικρατητικός χαρακτήρας Αυτοσωματικός επικρατητικός χαρακτήρας Χρωμοσώματα : Χρωμοσώματα : 19q13.2 →RPS19 19q13.2 →RPS19 10q22-23→RPS24 10q22-23→RPS24 15q25.2 →RPS17 15q25.2 →RPS17 RPS 5, RPL 11, RPL 35A RPS 5, RPL 11, RPL 35A Αδιευκρίνιστες περιπτώσεις Αδιευκρίνιστες περιπτώσεις

66
Επιπλοκές Ανάπτυξη ΜΔΣ, ΟΜΛ Ανάπτυξη ΜΔΣ, ΟΜΛ Ανάπτυξη μη αιματολογικών κακοηθειών (οστεοσάρκωμα) Ανάπτυξη μη αιματολογικών κακοηθειών (οστεοσάρκωμα) Απλαστική αναιμία Απλαστική αναιμία Μελέτες σε κυτταρικές καλλιέργειες ανέδειξαν βλάβη και στις τρεις κυτταρικές σειρές και όχι μόνο στην ερυθρά Μελέτες σε κυτταρικές καλλιέργειες ανέδειξαν βλάβη και στις τρεις κυτταρικές σειρές και όχι μόνο στην ερυθρά
 Ανάπτυξη μη αιματολογικών κακοηθειών (οστεοσάρκωμα) Απλαστική αναιμία Απλαστική αναιμία Μελέτες σε κυτταρικές καλλιέργειες ανέδειξαν βλάβη και στις τρεις κυτταρικές σειρές και όχι μόνο στην ερυθρά Μελέτες σε κυτταρικές καλλιέργειες ανέδειξαν βλάβη και στις τρεις κυτταρικές σειρές και όχι μόνο στην ερυθρά")
67
Παθογένεια Μελέτες in vitro ανέδειξαν διαταραχές στον πολ/μο, διαφοροποίηση, απόπτωση και την απάντηση σε κυτταροκίνες. Δεν αρκούν όμως για να ερμηνεύσουν την in vivo ανεπάρκεια της ερυθράς σειράς Μελέτες in vitro ανέδειξαν διαταραχές στον πολ/μο, διαφοροποίηση, απόπτωση και την απάντηση σε κυτταροκίνες. Δεν αρκούν όμως για να ερμηνεύσουν την in vivo ανεπάρκεια της ερυθράς σειράς Η πρόσφατη χαρτογράφηση 3 γονιδίων και η ανίχνευση των πρωτεϊνικών προϊόντων τους βοήθησε αρκετά στην κατανόηση της παθογένειάς της Η πρόσφατη χαρτογράφηση 3 γονιδίων και η ανίχνευση των πρωτεϊνικών προϊόντων τους βοήθησε αρκετά στην κατανόηση της παθογένειάς της

68
19q13.2 →RPS19 Χαρτογραφήθηκε το 1999 Χαρτογραφήθηκε το 1999 Κωδικοποιεί την RPS19 πρωτεΐνη (145 αα) συστατικό της 40s ριβοσωμικής υπομονάδας Κωδικοποιεί την RPS19 πρωτεΐνη (145 αα) συστατικό της 40s ριβοσωμικής υπομονάδας Ετεροζυγώτες για την συγκεκριμένη πρωτεΐνη μπορεί να είναι: Ετεροζυγώτες για την συγκεκριμένη πρωτεΐνη μπορεί να είναι: Πάσχοντες Πάσχοντες Συγγενείς πασχόντων χωρίς κλινινκές εκδηλώσεις οι οποίοι εμφανίζουν μεμονωμένη ↑e ADA Συγγενείς πασχόντων χωρίς κλινινκές εκδηλώσεις οι οποίοι εμφανίζουν μεμονωμένη ↑e ADA
 συστατικό της 40s ριβοσωμικής υπομονάδας Κωδικοποιεί την RPS19 πρωτεΐνη (145 αα) συστατικό της 40s ριβοσωμικής υπομονάδας Ετεροζυγώτες για την συγκεκριμένη πρωτεΐνη μπορεί να είναι: Ετεροζυγώτες για την συγκεκριμένη πρωτεΐνη μπορεί να είναι: Πάσχοντες Πάσχοντες Συγγενείς πασχόντων χωρίς κλινινκές εκδηλώσεις οι οποίοι εμφανίζουν μεμονωμένη ↑e ADA Συγγενείς πασχόντων χωρίς κλινινκές εκδηλώσεις οι οποίοι εμφανίζουν μεμονωμένη ↑e ADA")
69
8p23-22 → RPS24 8p23-22 → RPS24 Aφορά μικρό ποσοστό ασθενών (≈ 2%) Aφορά μικρό ποσοστό ασθενών (≈ 2%) Η RPS24 πρωτεΐνη συμμετέχει στη βιογένεση των ριβοσωμάτων Η RPS24 πρωτεΐνη συμμετέχει στη βιογένεση των ριβοσωμάτων 15q25.2 → RPS17 15q25.2 → RPS17 RPS17 πρωτεΐνη συμμετέχει στη ριβοσωμική λειτουργία RPS17 πρωτεΐνη συμμετέχει στη ριβοσωμική λειτουργία
 Aφορά μικρό ποσοστό ασθενών (≈ 2%) Η RPS24 πρωτεΐνη συμμετέχει στη βιογένεση των ριβοσωμάτων Η RPS24 πρωτεΐνη συμμετέχει στη βιογένεση των ριβοσωμάτων 15q25.2 → RPS17 15q25.2 → RPS17 RPS17 πρωτεΐνη συμμετέχει στη ριβοσωμική λειτουργία RPS17 πρωτεΐνη συμμετέχει στη ριβοσωμική λειτουργία")
70
Διάγνωση Οριστική διάγνωση είναι δυνατή για το 30% των ασθενών με βάση την ανίχνευση των 3 γνωστών μεταλλαγμένων γονιδίων

71
Θεραπεία Κορτικοστεροειδή Κορτικοστεροειδή Πρόγραμμα μεταγγίσεων για τους ανθεκτικούς στα κορτικοστεροειδή ασθενείς Πρόγραμμα μεταγγίσεων για τους ανθεκτικούς στα κορτικοστεροειδή ασθενείς Αλλογενής μεταμόσχευση μυελού οστών Αλλογενής μεταμόσχευση μυελού οστών

72
Συγγ. αμεγακαρυοκυτταρική θρομβοπενία Θρομβοπενία κατά τη βρεφική ηλικία χωρίς προσβολή των υπολοίπων σειρών Θρομβοπενία κατά τη βρεφική ηλικία χωρίς προσβολή των υπολοίπων σειρών Μυελόγραμμα: χαρακτηριστική απουσία ή περιορισμένη εκπροσώπιση της μεγακαρυοκυτταρικής σειράς Μυελόγραμμα: χαρακτηριστική απουσία ή περιορισμένη εκπροσώπιση της μεγακαρυοκυτταρικής σειράς Δεν συνοδεύεται από σωματικές ανωμαλίες Δεν συνοδεύεται από σωματικές ανωμαλίες 50% των ασθενών αναπτύσσουν ΑΑ σε ηλικία 5 ετών 50% των ασθενών αναπτύσσουν ΑΑ σε ηλικία 5 ετών

73
Χαρακτηρίζεται από γενετική ετερογένεια Χαρακτηρίζεται από γενετική ετερογένεια Αυτοσωμική υπολειπόμενη κληρονομικότητα σε μερικούς ασθενείς Αυτοσωμική υπολειπόμενη κληρονομικότητα σε μερικούς ασθενείς Μεταλλάξεις στο γονίδιο (1p34) που κωδικοποιεί τον υποδοχέα της θρομβοποιητίνης (c-mpl) Μεταλλάξεις στο γονίδιο (1p34) που κωδικοποιεί τον υποδοχέα της θρομβοποιητίνης (c-mpl) Ασθενείς με μετάλλαξη στο c-mpl γονίδιο μπορεί να εμφανίζουν διαταραχές και στις άλλες σειρές καθώς και στο ΚΝΣ Ασθενείς με μετάλλαξη στο c-mpl γονίδιο μπορεί να εμφανίζουν διαταραχές και στις άλλες σειρές καθώς και στο ΚΝΣ
 που κωδικοποιεί τον υποδοχέα της θρομβοποιητίνης (c-mpl) Μεταλλάξεις στο γονίδιο (1p34) που κωδικοποιεί τον υποδοχέα της θρομβοποιητίνης (c-mpl) Ασθενείς με μετάλλαξη στο c-mpl γονίδιο μπορεί να εμφανίζουν διαταραχές και στις άλλες σειρές καθώς και στο ΚΝΣ Ασθενείς με μετάλλαξη στο c-mpl γονίδιο μπορεί να εμφανίζουν διαταραχές και στις άλλες σειρές καθώς και στο ΚΝΣ")
74
Διάγνωση Αυξημένα επίπεδα ΤΡΟ ορού Αυξημένα επίπεδα ΤΡΟ ορού Ανίχνευση των μεταλλάξεων του υποδοχέα της ΤΡΟ Ανίχνευση των μεταλλάξεων του υποδοχέα της ΤΡΟ

75
Σχέση φαινότυπου/γονότυπου Τύπος Ι: πλήρης απώλεια της λειτουργικότητας του υποδοχέα της ΤΡΟ → πρώιμη εμφάνιση βαριάς θρομβοπενίας με εξέλιξη σε απλ. αναιμία, ΜΔΣ ή ΟΜΛ (2 έτη) Τύπος ΙΙ: μερική ανεπάρκεια του υποδοχέα ΤΡΟ → παροδική αύξηση των PLTs τα πρώτα χρόνια ζωής και καθυστερημένη εμφάνιση απλ. αναιμίας (5 έτη)
 Τύπος ΙΙ: μερική ανεπάρκεια του υποδοχέα ΤΡΟ → παροδική αύξηση των PLTs τα πρώτα χρόνια ζωής και καθυστερημένη εμφάνιση απλ. αναιμίας (5 έτη).")
76
Σ. TAR Θρομβοπενία από τη γέννηση και απουσία κερκίδας άμφω (αντίχειρες παρόντες δδ από FA) Θρομβοπενία από τη γέννηση και απουσία κερκίδας άμφω (αντίχειρες παρόντες δδ από FA) Αυξημένη ΤΡΟ πλάσματος με μειωμένη παρουσία ΜΓΚ στο μυελό Αυξημένη ΤΡΟ πλάσματος με μειωμένη παρουσία ΜΓΚ στο μυελό Κληρονομείται με αυτοσωματικό υπολειπόμενο χαρακτήρα – αναφορές και για επικρατητική κληρονομικότητα Κληρονομείται με αυτοσωματικό υπολειπόμενο χαρακτήρα – αναφορές και για επικρατητική κληρονομικότητα Η θρομβοπενία βελτιώνεται μετά τον 1 ο χρόνο ζωής (σπάνια ΟΛΛ, ΟΜΛ) Η θρομβοπενία βελτιώνεται μετά τον 1 ο χρόνο ζωής (σπάνια ΟΛΛ, ΟΜΛ)
 Θρομβοπενία από τη γέννηση και απουσία κερκίδας άμφω (αντίχειρες παρόντες δδ από FA) Αυξημένη ΤΡΟ πλάσματος με μειωμένη παρουσία ΜΓΚ στο μυελό Αυξημένη ΤΡΟ πλάσματος με μειωμένη παρουσία ΜΓΚ στο μυελό Κληρονομείται με αυτοσωματικό υπολειπόμενο χαρακτήρα – αναφορές και για επικρατητική κληρονομικότητα Κληρονομείται με αυτοσωματικό υπολειπόμενο χαρακτήρα – αναφορές και για επικρατητική κληρονομικότητα Η θρομβοπενία βελτιώνεται μετά τον 1 ο χρόνο ζωής (σπάνια ΟΛΛ, ΟΜΛ) Η θρομβοπενία βελτιώνεται μετά τον 1 ο χρόνο ζωής (σπάνια ΟΛΛ, ΟΜΛ).")
78
Βαρειά συγγενής ουδετεροπενία Χαρακτηρίζεται από: Χαρακτηρίζεται από: Διαταραχή ωρίμανσης της κοκκιώδους σειράς στο στάδιο προμυελο/μυελοκυττάρου Διαταραχή ωρίμανσης της κοκκιώδους σειράς στο στάδιο προμυελο/μυελοκυττάρου ΑΑΠ περιφ. αίματος < 0,5× 10 9/L ΑΑΠ περιφ. αίματος < 0,5× 10 9/L Συχνές βακτηριδιακές λοιμώξεις από την πρώτη βρεφική ηλικία Συχνές βακτηριδιακές λοιμώξεις από την πρώτη βρεφική ηλικία Θεωρείται προλευχαιμική κατάσταση (21% αναπτύσσουν λευχαιμία μετά από 10 έτη) Θεωρείται προλευχαιμική κατάσταση (21% αναπτύσσουν λευχαιμία μετά από 10 έτη)
 Θεωρείται προλευχαιμική κατάσταση (21% αναπτύσσουν λευχαιμία μετά από 10 έτη).")
79
Τύποι κληρονομικότητας Αυτοσωματικός επικρατών: Αυτοσωματικός επικρατών: Μεταλλάξεις στο γονίδιο της ELA2 (60% των περιπτώσεων). Υπόλοιπες περιπτώσεις είναι σποραδικές Μεταλλάξεις στο γονίδιο της ELA2 (60% των περιπτώσεων). Υπόλοιπες περιπτώσεις είναι σποραδικές Αυτοσωματικός υπολειπόμενος (Kostmann): Αυτοσωματικός υπολειπόμενος (Kostmann): Μεταλλάξεις στο γονίδιο HAX1 (30% περιπτώσεων) Μεταλλάξεις στο γονίδιο HAX1 (30% περιπτώσεων) Φυλοσύνδετος Φυλοσύνδετος Σποραδικές περιπτώσεις Σποραδικές περιπτώσεις
. Υπόλοιπες περιπτώσεις είναι σποραδικές Αυτοσωματικός υπολειπόμενος (Kostmann): Αυτοσωματικός υπολειπόμενος (Kostmann): Μεταλλάξεις στο γονίδιο HAX1 (30% περιπτώσεων) Μεταλλάξεις στο γονίδιο HAX1 (30% περιπτώσεων) Φυλοσύνδετος Φυλοσύνδετος Σποραδικές περιπτώσεις Σποραδικές περιπτώσεις.")
80
Παθοφυσιολογία Ι Μεταλλάξεις του ELA2 γονιδίου και του HAX1γονιδίου οδηγούν σε μειωμένη έκφραση των μεταγραφικών παραγόντων της κοκκιώδους σειράς μέσω καταστολής των αντίστοιχων γονιδίων (LEBF1) και σε πρώιμη απόπτωση των προγονικών κυττάρων Μεταλλάξεις του ELA2 γονιδίου και του HAX1γονιδίου οδηγούν σε μειωμένη έκφραση των μεταγραφικών παραγόντων της κοκκιώδους σειράς μέσω καταστολής των αντίστοιχων γονιδίων (LEBF1) και σε πρώιμη απόπτωση των προγονικών κυττάρων
 και σε πρώιμη απόπτωση των προγονικών κυττάρων Μεταλλάξεις του ELA2 γονιδίου και του HAX1γονιδίου οδηγούν σε μειωμένη έκφραση των μεταγραφικών παραγόντων της κοκκιώδους σειράς μέσω καταστολής των αντίστοιχων γονιδίων (LEBF1) και σε πρώιμη απόπτωση των προγονικών κυττάρων")
81
Ελαστάση 2 (ELA 2) Πρωτεάση σερίνης παραγόμενη στο στάδιο του προμυελοκυττάρου Πρωτεάση σερίνης παραγόμενη στο στάδιο του προμυελοκυττάρου Αποθηκεύεται στα πρωτογενή κοκκία των ουδετεροφίλων Αποθηκεύεται στα πρωτογενή κοκκία των ουδετεροφίλων Περισσότερες από 50 μεταλλάξεις έχουν περιγραφεί σε αυτοσ. επικρατείς και σποραδικές περιπτώσεις βαριάς συγγ. ουδετεροπενίας Περισσότερες από 50 μεταλλάξεις έχουν περιγραφεί σε αυτοσ. επικρατείς και σποραδικές περιπτώσεις βαριάς συγγ. ουδετεροπενίας

82
HAX 1 Μιτοχονδριακή πρωτεΐνη Μιτοχονδριακή πρωτεΐνη Αντιαποπτωτική δράση Αντιαποπτωτική δράση Μεταλλάξεις του HAX1 γονιδίου οδηγούν σε αδρανοποίηση της αντίστοιχης πρωτεΐνης, απώλεια του δυναμικού της μιτοχ. μεμβράνης και απελευθέρωση των προαποπτωτικών πρωτεϊνών με συνέπεια πρώιμη απόπτωση Μεταλλάξεις του HAX1 γονιδίου οδηγούν σε αδρανοποίηση της αντίστοιχης πρωτεΐνης, απώλεια του δυναμικού της μιτοχ. μεμβράνης και απελευθέρωση των προαποπτωτικών πρωτεϊνών με συνέπεια πρώιμη απόπτωση

83
Growth factor-independent protein 1 Μεταγραφική πρωτεΐνη Μεταγραφική πρωτεΐνη Μεταλλάξεις της οδηγούν σε: Μεταλλάξεις της οδηγούν σε: υπερέκφραση της ELA 2 και πρώιμη απόπτωση υπερέκφραση της ELA 2 και πρώιμη απόπτωση Υπερέκφραση του CSF-1 με αποτέλεσμα τη μετατροπή των προγονικών την κοκκιώδους σειράς σε μακροφάγα Υπερέκφραση του CSF-1 με αποτέλεσμα τη μετατροπή των προγονικών την κοκκιώδους σειράς σε μακροφάγα

84
WAS πρωτεΐνη Μεταλλάξεις στην WASp οδηγούν σε διαταραχές στη μίτωση με αποτέλεσμα μειωμένο πολλαπλασιασμό και αυξημένη απόπτωση των προγονικών κυττάρων της κοκκιώδους σειράς. Μεταλλάξεις στην WASp οδηγούν σε διαταραχές στη μίτωση με αποτέλεσμα μειωμένο πολλαπλασιασμό και αυξημένη απόπτωση των προγονικών κυττάρων της κοκκιώδους σειράς. Άλλες πρωτεΐνες: CD40 ligand, MAPBPIP, AP3B1, CHS1/LYST Άλλες πρωτεΐνες: CD40 ligand, MAPBPIP, AP3B1, CHS1/LYST

85
Παθοφυσιολογία ΙΙ (N Eng J Med 2009 Jan) Διαταραχές στη λειτουργία Glucose-6- phosphatase, catalytic subunit 3, σχετίζονται με σύνδρομο σοβαρής συγγ. ουδετεροπενίας συνοδευόμενο από ανωμαλίες της καρδιάς και του ουροποιογεννητικού συστήματος Διαταραχές στη λειτουργία Glucose-6- phosphatase, catalytic subunit 3, σχετίζονται με σύνδρομο σοβαρής συγγ. ουδετεροπενίας συνοδευόμενο από ανωμαλίες της καρδιάς και του ουροποιογεννητικού συστήματος

86
Παθοφυσιολογία ΙΙΙ Μηχανισμός λευχαιμογένεσης: Μηχανισμός λευχαιμογένεσης: Σχετίζεται με επίκτητες μεταλλάξεις του υποδοχέα του G-CSF Σχετίζεται με επίκτητες μεταλλάξεις του υποδοχέα του G-CSF 80% των ασθενών που αναπτύσσουν ΟΜΛ φέρουν τις συγκεκριμένες μεταλλάξεις 80% των ασθενών που αναπτύσσουν ΟΜΛ φέρουν τις συγκεκριμένες μεταλλάξεις

87
Διάγνωση Ιστορικό Ιστορικό Κλινική εικόνα Κλινική εικόνα ΑΑΠ < 500/μL ΑΑΠ < 500/μL Χαρακτηριστική εικόνα μυελογράμματος Χαρακτηριστική εικόνα μυελογράμματος Αναζήτηση συγκεκριμένων μεταλλάξεων (ELA2, HAX1) Αναζήτηση συγκεκριμένων μεταλλάξεων (ELA2, HAX1)
 Αναζήτηση συγκεκριμένων μεταλλάξεων (ELA2, HAX1)")
88
Θεραπεία Αντιμετώπιση λοιμώξεων Αντιμετώπιση λοιμώξεων Χορήγηση G-CSF Χορήγηση G-CSF ΜΜΟ ΜΜΟ

89
Συμπερασματικά... Τα ΣΚΜΑ είναι σπάνια και χωρίς θεραπεία θανατηφόρα Τα ΣΚΜΑ είναι σπάνια και χωρίς θεραπεία θανατηφόρα Η κλινική εικόνα ποικίλει τόσο ως προς τη βαρύτητα όσο και την ηλικία πρώτης εκδήλωσης Η κλινική εικόνα ποικίλει τόσο ως προς τη βαρύτητα όσο και την ηλικία πρώτης εκδήλωσης Νεαροί ενήλικες με επιθηλιακούς καρκίνους μπορεί να πάσχουν από ΣΚΜΑ Νεαροί ενήλικες με επιθηλιακούς καρκίνους μπορεί να πάσχουν από ΣΚΜΑ

90
Συμπερασματικά... Τα τελευταία 10 χρόνια έγιναν σημαντικά βήματα στην κατανόηση της μοριακής βάσης των ΣΚΜΑ που βοηθούν στη διάγνωση όταν η εικόνα δεν είναι τυπική Τα τελευταία 10 χρόνια έγιναν σημαντικά βήματα στην κατανόηση της μοριακής βάσης των ΣΚΜΑ που βοηθούν στη διάγνωση όταν η εικόνα δεν είναι τυπική Η επιτυχής ΜΜΟ λύνει το αιματολογικό πρόβλημα αλλά δεν απομακρύνει τον κίνδυνο κακοηθειών... Η επιτυχής ΜΜΟ λύνει το αιματολογικό πρόβλημα αλλά δεν απομακρύνει τον κίνδυνο κακοηθειών...

Παρόμοιες παρουσιάσεις












 8 η παράδοση.>")






 Κύτταρο Β ) Δομές DNA - RNA Παρουσίαση Βιολογίας.>")



