Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
Κεφάλαιο 9 Βιοχημική γενετική

2
9.1 – 9.3

3
9.1 Συγγενείς διαταραχές του μεταβολισμού

4
Συγγενείς διαταραχές του μεταβολισμού
Συγγενής νόσος : πρόκειται για κάθε νόσο που υφίσταται κατά τη γέννηση σε αντίθεση με το επίκτητο. Ένα συγγενές γνώρισμα ή νόσος μπορεί να είναι κληρονομικό , αλλά μπορεί να οφείλεται σε κάποιαν επίδραση επί του εμβρύου κατά την διάρκεια της ενδομητρίου ζωής.

5
Συγγενείς διαταραχές του μεταβολισμού
Οι διαταραχές του μεταβολισμού ομαδοποιούνται σε κατηγορίες που αφορούν τη λειτουργικότητα ενός ενζύμου ή μιας πρωτεΐνης , το μεταβολίτη, τη μεταβολική οδό ή το κυτταρικό οργανίδιο. Έχουν περιγραφεί περίπου 200 διαταραχές αυτού του τύπου. Οι περισσότερες διαταραχές αυτού του τύπου κληρονομούνται με αυτοσωμικά υπολειπόμενα ή Χ-φυλοσύνδετα υπολειπόμενα γονίδια και μόνο λίγες με αυτοσωμικά επικρατή. Αρχικά ο Garrod και αργότερα οι Beadle και Tatum ανέπτυξαν την ιδίέα οτι οι μεταβολικές οδοί στον άνθρωπο και άλλους οργανισμούς , ακολουθούν ορισμένα στάδια. Πρότειναν οτι κάθε στάδιο ελέγχεται από ένα ένζυμο που είναι προϊόν ενός ορισμένου γονιδίου.

10
Γιατί περισσότερες διαταραχές του μεταβολισμού κληρονομούνται με υπολειπόμενο τύπο κληρονόμησης (αυτοσωμικό ή φυλοσύνδετο) ; Γενικά ισχύει ότι τα μεταλλαγμένα γονίδια που κωδικοποιούν ένζυμα κατά κανόνα ακολουθούν υπολειπόμενο τύπο κληρονόμησης. Η ερμηνεία έχει ως εξής : Τα δύο ομόλογα γονίδια που κωδικοποιούν ένα ένζυμο είναι υπεύθυνα για μια συγκεκριμένη κυτταρική λειτουργία. Θα μπορούσαμε να τα παρομοιάσουμε τα γονίδια των ενζύμων με εργάτες σε μια οικοδομή αναλαμβάνουν ανά δύο μια συγκεκριμένη εργασία. Αν ο ένας από τους δύο εργαζόμενους αρνείται να εργαστεί ο δεύτερος θα δουλέψει κανονικά και η εργασία αυτή θα ολοκληρωθεί ( ίσως ο δεύτερος εργάτης δουλέψει περισσότερο και ίσως το τελικό αποτέλεσμα να έχει μερικές ατέλειες , γενικά όμως η δουλειά θα γίνει). Ομοίως και για τα δύο αλληλόμορφα γονίδια αν το ένα που έχει κληρονομηθεί είναι ελαττωματικό τότε το δεύτερο γονίδιο θα εκάνει την ίδια εργασία (ίσως υπερεκφραστεί και ίσως η ποιότητα του αποτελέσματος να μην είναι η ίδια). Επομένως τα γονίδια που εκφράζουν λειτουργικές πρωτείνες γενικά είναι υπολειπόμενα καθώς δεν αρκεί ένα μεταλλαγμένο γονίδιο για να έχουμε μεταλλαγμένο φαινότυπο.
. Ομοίως και για τα δύο αλληλόμορφα γονίδια αν το ένα που έχει κληρονομηθεί είναι ελαττωματικό τότε το δεύτερο γονίδιο θα εκάνει την ίδια εργασία (ίσως υπερεκφραστεί και ίσως η ποιότητα του αποτελέσματος να μην είναι η ίδια). Επομένως τα γονίδια που εκφράζουν λειτουργικές πρωτείνες γενικά είναι υπολειπόμενα καθώς δεν αρκεί ένα μεταλλαγμένο γονίδιο για να έχουμε μεταλλαγμένο φαινότυπο.")
11
Επικρατής κληρονόμηση
Αντίθετα ένα γονίδιο που κωδικοποιεί πρωτεΐνη που συμμετέχει σε δομικές αλληλεπιδράσεις (πχ ομοδιμερισμός) όταν εμφανιστεί μεταλλαγμένο θα αλληλεπιδράσει με το φυσιολογικό προϊόν του δεύτερου αλληλομόρφου και ενδεχομένως να καταστείλει την λειτουργία του (επικρατές πρότυπο κληρονόμησης) όπως ακριβώς όταν ένας εργάτης όχι μόνο δεν εργάζεται αλλά "αλληλεπιδρά" και με τους άλλους εργάτες και δεν τους αφήνει να εργαστούν
 όταν εμφανιστεί μεταλλαγμένο θα αλληλεπιδράσει με το φυσιολογικό προϊόν του δεύτερου αλληλομόρφου και ενδεχομένως να καταστείλει την λειτουργία του (επικρατές πρότυπο κληρονόμησης) όπως ακριβώς όταν ένας εργάτης όχι μόνο δεν εργάζεται αλλά αλληλεπιδρά και με τους άλλους εργάτες και δεν τους αφήνει να εργαστούν.")
12
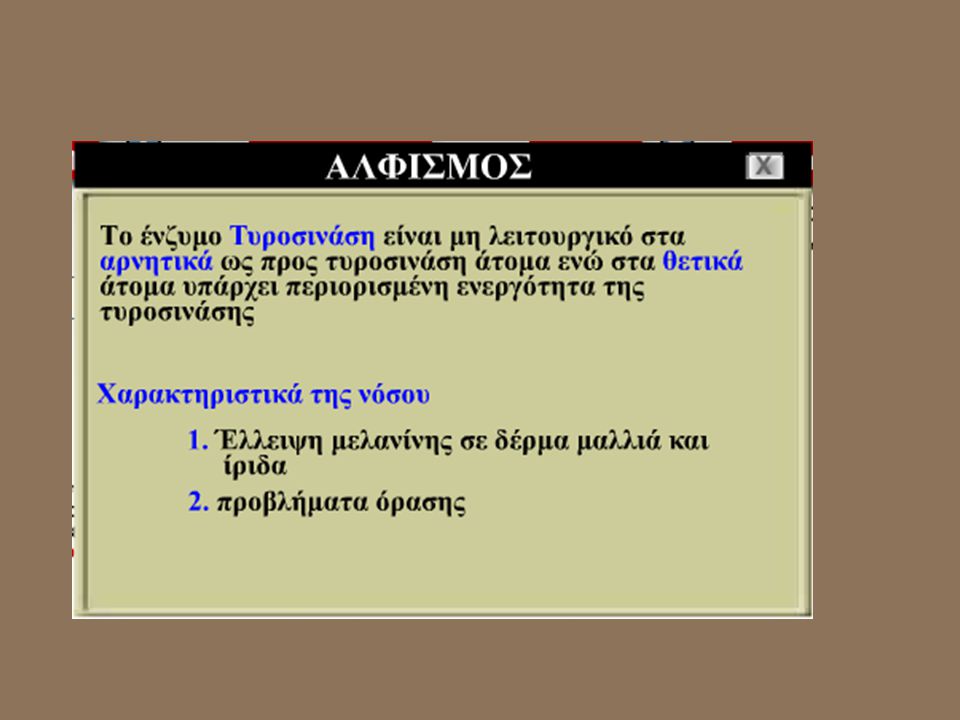
9.2 Διαταραχές μεταβολισμού τύπου Garrod
Διαδραστική εικόνα

16
9.3 Λοιπές διαταραχές

17
Διαταραχές μεταφοράς αμινοξέων
Κυστινουρία Τα άτοµα µε κυστινουρία εµφανίζουν συµπτώµατα λόγω της δηµιουργίας λίθων στους νεφρούς οι οποίοι συνήθως αποτελούνται από κυστίνη και εµφανίζονται σε διάφορες ηλικίες. Η διαταραχή προκαλείται από την ελαττωµένη επαναπορρόφηση από τα νεφρικά σωληνάρια τεσσάρων αµινοξέων ( κυστίνη, λυσίνη, ορνιθίνη, αργινίνη ).
.")
18
Διαταραχές μεταφοράς αμινοξέων
Θεραπεία συνίσταται στη χορήγηση D-πενικιλαµίνης, η οποία αντιδρά µε την κυστίνη και δηµιουργεί ένα σύµπολο πιο ευδιάλυτο από την ελεύθερη κυστίνη και δηµιουργεί ένα πιο ευδιάλυτο σύµπλοκο.

19
Διαταρχές κύκλου ουρίας
Έλλειψη τρανσκαρβαµυλάσης της ορνιθίνης Ο κύκλος της ουρίας φυσιολογικά µεταβολίζει την αµµωνία προς τον σχηµατισµό ουρίας και η τρανσκαρβαµυλάση της ορνιθίνης είναι ένα από τα σηµαντικότερα ένζυµα του κύκλου. Η έλλειψη τρανσκαρβαµυλάσης της ορνιθίνης σηµαίνει συσσώρευση αµµωνίας η οποία είναι τοξική, ιδιαίτερα γιατο νευρικό σύστηµα. Εκδηλώνεται στο νεογνικό στάδιο µε συµπτώµατα όπως δυσκολία Στη διατροφή και εµφάνιση λήθαργου που γρήγορα οδηγούν σε κώµα και τελικώς σε θάνατο από τον πρώτο µήνα της ζωής. Τα άτοµα που νοσούν είναι κατά κανόνα αρσενικά εφόσον η διαταραχή είναι φυλοσύνδετη .

20
Διαταραχές υδατανθράκων
Γαλακτοζαιµία Η γαλακτόζη µεταβολίζεται φυσιολογικά µε τη συµµετοχή του ενζύµου τρανσφεράση της 1-Ρ-ουριδυλ-γαλακτόζης. Η έλλειψη του ενζύµου αυτού µε ταυτόχρονη κατανάλωση τροφών που περιέχουν γαλακτόζη (ή λακτόζη) προκαλεί στο νεογνό εµετούς και αργότερα προκαλεί : • ∆ιανοητική καθυστέρηση • Καταρράκτη • Κίρρωση του ήπατος Αντιµετώπιση : Πρώιµη διάγνωση και εφαρµογή δίαιτας µε υποκατάστατα του γάλακτος που δεν περιέχουν γαλακτόζη ή λακτόζη
 προκαλεί. στο νεογνό εµετούς και αργότερα προκαλεί : • ∆ιανοητική καθυστέρηση. • Καταρράκτη. • Κίρρωση του ήπατος. Αντιµετώπιση : Πρώιµη διάγνωση και εφαρµογή δίαιτας µε υποκατάστατα του γάλακτος που δεν περιέχουν γαλακτόζη ή λακτόζη.")
21
Διαταραχές υδατανθράκων
Ασθένειες αποθήκευσης γλυκογόνου Σ΄αυτές τις ασθένειες το γλυκογόνο συσσωρεύεται στους σκελετικούς και καρδιακούς µυς και το ήπαρ. Το γλυκογόνο πια όχι µόνο συσσωρεύεται επικίνδυνα αλλά δεν µπορεί πια να είναι φυσιολογική πηγή γλυκόζης εφόσον δεν λειτουργεί κάποιο ένζυµο του µεταβολισµού του. Έτσι το άτοµο αντιµετωπίζει υπογλυκαιµία, νευρολογικές διαταραχές και δυσλειτουργία του ήπατος.

22
Διαταραχές υδατανθράκων
Ασθένεια Mc Ardle Ανεπάρκεια φωσφορυλάσης του γλυκογόνου ( της ειδικής για τους µυς) προκαλεί επώδυνες µυικές συσπάσεις. ∆εν υπάρχει δραστική θεραπεία
 προκαλεί επώδυνες µυικές συσπάσεις. ∆εν υπάρχει δραστική θεραπεία.")
23
Διαταραχές υδατανθράκων
Ασθένεια Pompe Ανεπάρκεια της α-γλυκοσιδάσης των λυσοσωµάτων (απαραίτητο για την διάσπαση του γλυκογόνου) προκαλεί καρδιακή ανεπάρκεια και θάνατο από το πρώτο έτος ζωής. ∆εν υπάρχει δραστική θεραπεία
 προκαλεί καρδιακή ανεπάρκεια και θάνατο από το πρώτο έτος ζωής. ∆εν υπάρχει δραστική θεραπεία.")
24
Διαταραχές μεταβολισμού λιποπρωτεΐνών

25
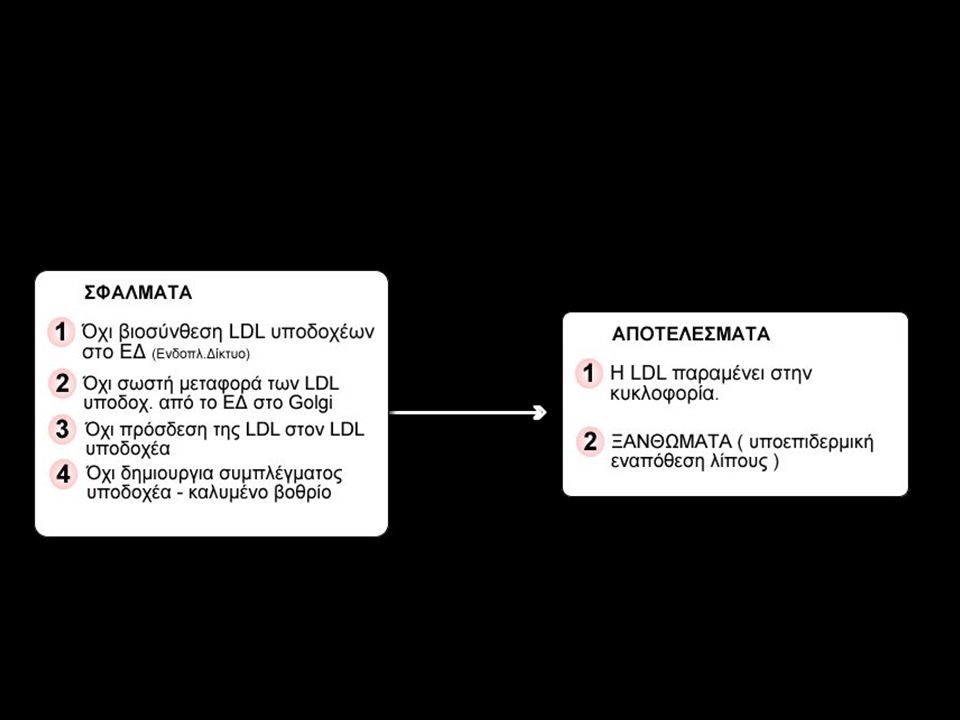
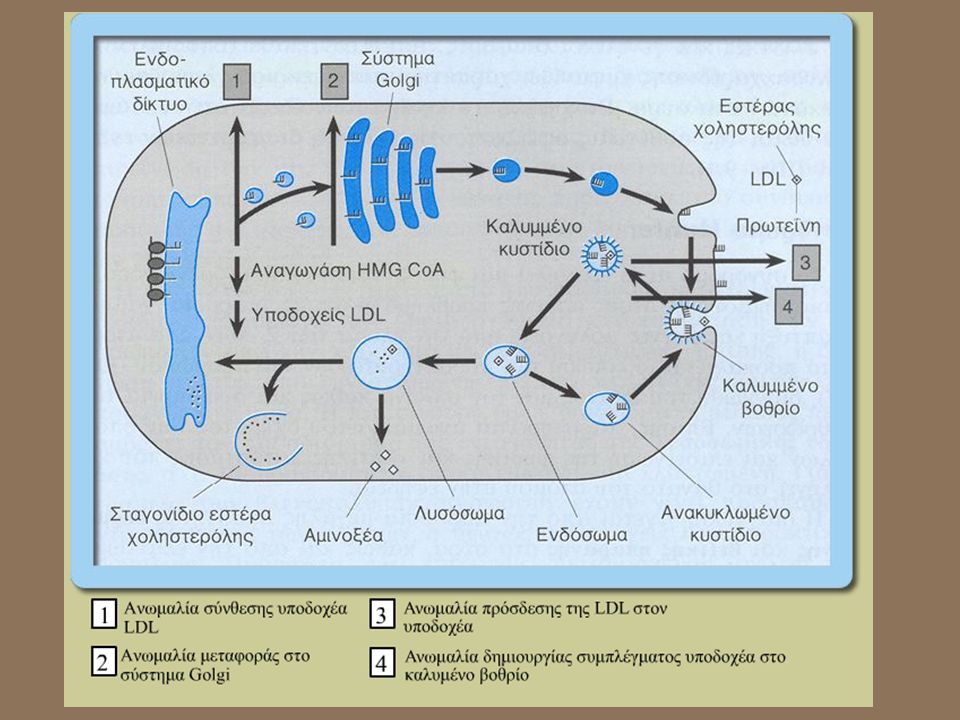
Οικογενής υπερχολυστεροναιµία
Πρόκειται για ιδιόµορφη µορφή αυτοσωµικής επικρατούς διατραχής ! που αν και επικρατής τα ετερόζυγα άτοµα Αα έχουν λιγότερα συµπτώµατα από τα οµόζυγα άτοµα ΑΑ. Τα ηπατοκύτταρα έχουν ανάγκη χοληστερόλης και είτε την βιοσυνθέτουν είτε την προσλαµβάνουν εξωτερικά µέσω των υποδοχέων LDL (χαµηλής πυκνότητας λιποπρωτεΐνη ). Τα υψηλά επίπεδα χολυστερόλης στο αίµα ατόµων που πάσχουν από υπερχολυστεροναιµία προέρχονται από ελαττωµάτική σύνθεση του υποδοχέα της LDL χολυστερόλης.
. Τα υψηλά επίπεδα χολυστερόλης στο αίµα ατόµων που πάσχουν από υπερχολυστεροναιµία προέρχονται από ελαττωµάτική σύνθεση του υποδοχέα της LDL χολυστερόλης.")
28
Animation για υπερχολυστεροναιμία από www.sumanasinc.com

29
Λυσσοσωμικές ασθένειες συσσώρευσης

30
Λυσσοσωμικές ασθένειες συσσώρευσης
Τα βιολογικά µακροµόρια φυσιολογικά βρίσκονται σε µια λεπτή ισορροπία σύνθεσης και αποικοδόµησης. Τα λυσσοσώµατα φυσιολογικά συµµετέχουν στην διάσπαση των µακροµορίων. Ελλείψεις σε κάποιο λυσοσωµικό ένζυµο είναι πιθανόν να προκαλέσει συσσώρευση κάποιου µακροµορίου. Τα παιδιά που εµφανίζουν αυτές τις ασθένειες είναι φυσιολογικά κατά την γέννηση ενώ τα συµπτώµατα εµφανίζονται σε µεγαλύτερη ηλικία

31
Λυσσοσωμικές ασθένειες συσσώρευσης
Βλεννοπολυσακχαριδώσεις Πρόκειται για µια οµάδα 12 νόσων που στα πάσχοντα άτοµα προκαλείται ελαττωµατική διάσπαση των υδατανθράκων ( ελαττωµατική διάσπαση των πλευρικών αλυσίδων των υδατανθράκων των οξέων των πολυσακχαριτών ). Η συσσώρευση των πολυσακχαριτών προκελαί προβλήµατα στο σκελετικό σύστηµα, στο αγγειακό σύστηµα και το νευρικό σύστηµα. Για κάθε βλεννοπολυσακχαρίδωση υπάρχει χαρακτηριστική έκκριση γλυκοζαµινογλυκάνων στα ούρα. Σύνδροµο Hunter (ελαττωµένη ενεργότητα µιας λυσοσωµικής υδρολάσης ) Σύνδρομο Hurler ( η σημαντικότερη νόσος των βλεννοπολυσακχαριδώσεων)
. Η συσσώρευση των πολυσακχαριτών προκελαί προβλήµατα στο σκελετικό σύστηµα, στο αγγειακό σύστηµα και το νευρικό σύστηµα. Για κάθε βλεννοπολυσακχαρίδωση υπάρχει χαρακτηριστική έκκριση γλυκοζαµινογλυκάνων στα ούρα. Σύνδροµο Hunter (ελαττωµένη ενεργότητα µιας λυσοσωµικής υδρολάσης ) Σύνδρομο Hurler ( η σημαντικότερη νόσος των βλεννοπολυσακχαριδώσεων)")
32
Σφιγγολιπιδώσεις Ανικανότητα διάσπασης των σφιγγολιπιίδων τα
Σταδιακά προκαλούνται σπασµοί , επιληψία, και Θάνατος κατά την παιδική ηλικία Νόσος Tay- Sachs (Εβραίοι Ασκενάζι) Νόσος Gaucher (Εβραίοι Ασκενάζι)
 Νόσος Gaucher (Εβραίοι Ασκενάζι)")
33
Ashkenazi (Από εβραϊκό Ashkenaz, " Γερμανία"), πληθυντικός Ashkenazim, οποιοιδήποτε από τους Εβραίους που έζησαν στην κοιλάδα της Ρηνανίας και στη γειτονική Γαλλία πριν από τη μετανάστευσή τους ανατολικά στα σλαβικά εδάφη (π.χ., Πολωνία, Λιθουανία, Ρωσία) μετά από τις σταυροφορίες (11ος –13ος αιώνας). Μετά από τις διώξεις 17ος-αιώνα στην Ανατολική Ευρώπη, οι μεγάλοι αριθμοί αυτών των Εβραίων επανεγκαταστήθηκαν στη δυτική Ευρώπη, όπου αφομοιώθηκαν, όπως είχαν κάνει στην Ανατολική Ευρώπη, με άλλες εβραϊκές κοινότητες. Εγκαίρως, όλοι οι Εβραίοι που είχαν υιοθετήσει το τελετουργικό συναγωγών "γερμανικής ιεροτελεστίας" αναφέρθηκαν ως Ashkenazim για να τους διακρίνουν από (ισπανική ιεροτελεστία) τους Εβραίους Sephardic. Το Ashkenazim διαφέρει από Sephardim στην προφορά των εβραϊκών τους, στις πολιτιστικές παραδόσεις, και ειδικά στη λειτουργία συναγωγών. Σήμερα οι Ashkenazim αποτελούν περισσότερο από 80 τοις εκατό όλου του εβραϊκού πληθυσμού στον κόσμο, ξεπερνώντας αριθμητικώς απέραντα τους Εβραίους Sephardic. Στο 20ο αιώνα , οι Εβραίοι Ashkenazic αρίθμησαν περισσότερο από Στο Ισραήλ οι αριθμοί Ashkenazim και Sephardim είναι κατά προσέγγιση ίσοι.
, πληθυντικός Ashkenazim, οποιοιδήποτε από τους Εβραίους που έζησαν στην κοιλάδα της Ρηνανίας και στη γειτονική Γαλλία πριν από τη μετανάστευσή τους ανατολικά στα σλαβικά εδάφη (π.χ., Πολωνία, Λιθουανία, Ρωσία) μετά από τις σταυροφορίες (11ος –13ος αιώνας). Μετά από τις διώξεις 17ος-αιώνα στην Ανατολική Ευρώπη, οι μεγάλοι αριθμοί αυτών των Εβραίων επανεγκαταστήθηκαν στη δυτική Ευρώπη, όπου αφομοιώθηκαν, όπως είχαν κάνει στην Ανατολική Ευρώπη, με άλλες εβραϊκές κοινότητες. Εγκαίρως, όλοι οι Εβραίοι που είχαν υιοθετήσει το τελετουργικό συναγωγών γερμανικής ιεροτελεστίας αναφέρθηκαν ως Ashkenazim για να τους διακρίνουν από (ισπανική ιεροτελεστία) τους Εβραίους Sephardic. Το Ashkenazim διαφέρει από Sephardim στην προφορά των εβραϊκών τους, στις πολιτιστικές παραδόσεις, και ειδικά στη λειτουργία συναγωγών. Σήμερα οι Ashkenazim αποτελούν περισσότερο από 80 τοις εκατό όλου του εβραϊκού πληθυσμού στον κόσμο, ξεπερνώντας αριθμητικώς απέραντα τους Εβραίους Sephardic. Στο 20ο αιώνα , οι Εβραίοι Ashkenazic αρίθμησαν περισσότερο από Στο Ισραήλ οι αριθμοί Ashkenazim και Sephardim είναι κατά προσέγγιση ίσοι.")
34
Διαταραχές μεταβολισμού πουρίνης πυριμιδίνης
Σύνδοµο Lesch – Nyhan Κληρονοµική µεταβολική αναταραχή που έχει επιπτώσεις στο κεντρικό νευρικό σύστηµα και πουχαρακτηρίζεται από διανοητική καθυστέρηση και επιθετική συµπεριφορά,. Η αιτία του συνδρόµου είναι ένα ελαττωµατικό ένζυµο το οποίο φυσιολογικά είναι ιδιαίτερα ενεργό στα κύτταρα του εγκεφάλου και συµµετέχει στο µεταβολισµό των πουρινών. Το σύνδροµο διαβιβάζεται από ένα υπολειπόµενο φυλοσύνδετο γονίδιο, προσβάλλοντας κυρίως αρσενικά άτοµα και είναι δυνατό να ανιχνευθεί πριν από τη γέννηση όταν οι γονείς είναι φορείς.

35
Λοιπές διαταραχές ∆ιαταραχές οργανικών οξέων
∆ιαταραχές µεταβολισµού πορφυρινών ( Η αίµη είναι µια πορφυρίνη) ∆ιαταραχές του µεταβολισµού του χαλκού Νόσος Μenke Nόσος Wilson
 ∆ιαταραχές του µεταβολισµού του χαλκού. Νόσος Μenke. Nόσος Wilson.")
36
Λοιπές διαταραχές ∆ιαταραχές βιοσύνθεσης των ορµονών του θυροειδούς
Συγγενής έλλειψη θυροξίνης Η θυροξίνη παίζει σπουδαίο ρόλο στην υγιή ανάπτυξη του παιδιού απουσία θυροξίνης επέρχεται διανοητική καθυστέρηση (κρετινισµός). Τα συµπτώµατα αντιµετωπίζονται µε χορήγηση θυροξίνης.
. Τα συµπτώµατα αντιµετωπίζονται µε χορήγηση θυροξίνης.")
37
Λοιπές διαταραχές ∆ιαταραχές υπεροξεισωµάτων
Τα υπεροξεισώµατα βρίσκονται σε µεγάλη συγκέτρωση στο ήπαρ και στα νεφρά. Συµµετέχουν στην οξείδωση των λιπαρών οξέων και την βιοσύνθεση της χοληστερόλης. (α) ∆ιαταραχές βιογένεσης υπεροξεισωµάτων Σύνδροµο Zellweger ( Θάνατος πριν συµπληρώσουν το πρώτο έτος ζωής ) (β) ∆ιαταραχές των ενζύµων των υπεροξεισωµάτων Αδρενογλευκοδυστροφία
 ∆ιαταραχές βιογένεσης υπεροξεισωµάτων. Σύνδροµο Zellweger ( Θάνατος πριν συµπληρώσουν το πρώτο έτος ζωής ) (β) ∆ιαταραχές των ενζύµων των υπεροξεισωµάτων. Αδρενογλευκοδυστροφία.")
Παρόμοιες παρουσιάσεις






















>")


