Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

1
ΠΡΟΛΗΨΗ ΓΕΝΕΤΙΚΩΝ ΝΟΣΗΜΑΤΩΝ

2
Carrod αρχές του 20ου αιώνα και αφορά μια ασθένεια του μεταβολισμού, την αλκαπτονουρία. Τα άτομα αυτά εξαιτίας μιας αυτοσωμικής μετάλλαξης δεν μπορούν να παραγάγουν ένα ένζυμο που διασπά το αμινοξύ φαινυλαλανίνη, μια ουσία που βρίσκεται άφθονη στο ψωμί, το τυρί, τα αυγά, τα ψάρια και το γάλα. Αυτό έχει σαν αποτέλεσμα τη συγκέντρωση της ουσίας σε υψηλά επίπεδα στο αίμα και την αποβολή της σε μεγάλες ποσότητες στα ούρα (φαινυλκετονουρία -ΡΚU- ή αλκαπτονουρία). Παράλληλα, η μεγάλη συγκέντρωση της στο αίμα προξενεί βαριά πνευματική καθυστέρηση. Επιβολή αυστηρής δίαιτας η οποία αποτελείται απο λαχανικά και φρούτα, καθώς και ένα τεχνικό μίγμα πρωτεϊνών χωρίς αυτό το αμινοξύ. ΠΡΟΪΟΝΤΑ LIGHT? Αλκαπτονουρία
. Παράλληλα, η μεγάλη συγκέντρωση της στο αίμα προξενεί βαριά πνευματική καθυστέρηση. Επιβολή αυστηρής δίαιτας η οποία αποτελείται απο λαχανικά και φρούτα, καθώς και ένα τεχνικό μίγμα πρωτεϊνών χωρίς αυτό το αμινοξύ. ΠΡΟΪΟΝΤΑ LIGHT. Αλκαπτονουρία.")
3
Ανάλογη περίπτωση με την ΡΚU είναι αυτή της γαλακτοσαιμίας: Στην περίπτωση αυτή, τα βρέφη που γεννιούνται με την πάθηση στερούνται ενός ενζύμου που διασπά τη γαλακτόζη, η οποία είναι το σάκχαρο του γάλατος. Και εδώ η πάθηση οφείλεται σε αυτοσωμικό υπολειπόμενο γονίδιο. Και στην περίπτωση αυτή παρατηρείται συγκέντρωση κάποιου τοξικού μεταβολίτη στο αίμα (γενετικά νοσήματα του μεταβολισμού), καθώς και υψηλή αποβολή του στα ούρα. Παράλληλα, η πάθηση συνοδεύεται από μια σειρά βαριά συμπτώματα, όπως πνευματική και σωματική καθυστέρηση, και πρόωρο θάνατο. Κι εδώ η αντιμετώπιση γίνεται με διαιτητικό τρόπο κι έτσι το γάλα αντικαθίσταται από υποκατάστατα χωρίς γαλακτόζη. Γαλακτοσαιμία
, καθώς και υψηλή αποβολή του στα ούρα. Παράλληλα, η πάθηση συνοδεύεται από μια σειρά βαριά συμπτώματα, όπως πνευματική και σωματική καθυστέρηση, και πρόωρο θάνατο. Κι εδώ η αντιμετώπιση γίνεται με διαιτητικό τρόπο κι έτσι το γάλα αντικαθίσταται από υποκατάστατα χωρίς γαλακτόζη. Γαλακτοσαιμία.")
4
Κυστική ίνωση Είναι μια σοβαρή γενετική νόσος, η πιο συχνή υποτελής αυτοσωμική διαταραχή στα λευκά (Καυκάσια) παιδιά.Τα κύρια συμπτώματα οφείλονται σε διαταραχή των εξωκρινών εκκρίσεων. Κατά κύριο λόγο προσβάλλει τα πνευμόνια και το πάγκρεας, προκαλεί συχνές λοιμώξεις του αναπνευστικού συστήματος και ελαττωμένη απορρόφηση της τροφής. Η λεπτή ινώδης βλέννη που παράγεται από τους βρόγχους, είναι συνήθως επικίνδυνη γιατί είναι δύσκολο να αποβληθεί με το βήξιμο, έτσι παρατηρούνται συχνές λοιμώξεις του αναπνευστικού συστήματος, όπως είναι η πνευμονία. Κάθε κρίση της λοίμωξης αφήνει τα πνευμόνια ελαφρά πιο πειραγμένα από πριν και έτσι η υγεία του παιδιού χειροτερεύει. Η θεραπεία με αντιβιοτικά και η έντονη φυσικοθεραπεία δεν είναι δυνατόν να αποτρέψει τέτοια επεισόδια αλλά βοηθά να αντιμετωπισθούν. Το πάγκρεας μπλοκάρεται από ινώδεις εκκρίσεις και έτσι δεν μπορεί να παράγει πεπτικούς χυμούς σε επαρκή ποσότητα. Έτσι το παιδί παρουσιάζει χρόνια διάρροια, αδυνατίζει και είναι καχεκτικό. Οι άνδρες δεν είναι γόνιμοι εξαιτίας μη φυσιολογικών εκκρίσεων βλέννης στο σπερματικό πόρο. Η απώλεια χλωριούχων ιόντων με τον ιδρώτα είναι δυνατό να είναι τόσο σοβαρή, ώστε να προκαλέσει πλήρη κατάρρευση, όταν ο καιρός είναι ζεστός. Στο Ενωμένο Βασίλειο, η κυστική ίνωση προσβάλλει περίπου ένα παιδί κάθε 2.000 γεννήσεις. Έχει εκτιμηθεί ότι περίπου 1 στα 20 άτομα είναι ετερόζυγος φορέας του γονιδίου της κυστικής ίνωσης. Ο κίνδυνος ότι ένας φορέας θα ζευγαρώσει με έναν άλλο φορέα είναι περίπου 1 στις 400. Στην Ελλάδα η κυστική ίνωση προσβάλλει 1 παιδί κάθε 20.000 γεννήσεις.

5
Η μυϊκή δυστροφία καλύπτει μια ομάδα κληρονομικών διαταραχών στις οποίες εμφανίζεται αργός αλλά προοδευτικός εκφυλισμός των μυών, συνήθως στους μηρούς, τους ώμους και τις γάμπες. Η μυϊκή δυστροφία Duchenne (DMD) εμφανίζεται νωρίς, στην παιδική ηλικία. Προσβάλλει το παιδικό σώμα και συχνά εντοπίζεται την στιγμή που το παιδί αρχίζει να περπατά. Με την πάροδο του χρόνου εξελίσσεται και, όταν το παιδί φθάσει στην ηλικία των 10 χρόνων περιορίζεται σε αναπηρική καρέκλα. Ο θάνατος επέρχεται στην εφηβεία, έτσι, οι άρρωστοι δεν ζουν πέραν των 20 χρόνων. Αυτό το υπολειπόμενο γονίδιο εντοπίζεται στον κοντό βραχίονα του χρωμοσώματος Χ. Η φύση της αρρώστιας είναι τέτοια, που το γονίδιο δεν είναι δυνατόν να μεταβιβασθεί από τους άνδρες, γιατί αυτοί προσβάλλονται και πεθαίνουν πριν φθάσουν στην αναπαραγωγική ηλικία. Μεταβιβάζεται από φορείς (ετερόζυγες) γυναίκες, που πολύ σπάνια εμφανίζουν κάποιο από τα συμπτώματα της αρρώστιας. Η DMD έχει από τις μεγαλύτερες ταχύτητες μεταλλάξεων που έχουν μετρηθεί στον άνθρωπο, είναι περίπου 1 στις 10.000. Δεν υπάρχει θεραπεία, αλλά τώρα είναι πια δυνατή η προγεννητική διάγνωση και ο εντοπισμός των φορέων (ετερόζυγοι). Υπάρχουν μελέτες με βλαστοκύτταρα που δίνουν ελπίδες. Μυϊκή Δυστροφία Duchenne
 εμφανίζεται νωρίς, στην παιδική ηλικία. Προσβάλλει το παιδικό σώμα και συχνά εντοπίζεται την στιγμή που το παιδί αρχίζει να περπατά. Με την πάροδο του χρόνου εξελίσσεται και, όταν το παιδί φθάσει στην ηλικία των 10 χρόνων περιορίζεται σε αναπηρική καρέκλα. Ο θάνατος επέρχεται στην εφηβεία, έτσι, οι άρρωστοι δεν ζουν πέραν των 20 χρόνων. Αυτό το υπολειπόμενο γονίδιο εντοπίζεται στον κοντό βραχίονα του χρωμοσώματος Χ. Η φύση της αρρώστιας είναι τέτοια, που το γονίδιο δεν είναι δυνατόν να μεταβιβασθεί από τους άνδρες, γιατί αυτοί προσβάλλονται και πεθαίνουν πριν φθάσουν στην αναπαραγωγική ηλικία. Μεταβιβάζεται από φορείς (ετερόζυγες) γυναίκες, που πολύ σπάνια εμφανίζουν κάποιο από τα συμπτώματα της αρρώστιας. Η DMD έχει από τις μεγαλύτερες ταχύτητες μεταλλάξεων που έχουν μετρηθεί στον άνθρωπο, είναι περίπου 1 στις Δεν υπάρχει θεραπεία, αλλά τώρα είναι πια δυνατή η προγεννητική διάγνωση και ο εντοπισμός των φορέων (ετερόζυγοι). Υπάρχουν μελέτες με βλαστοκύτταρα που δίνουν ελπίδες. Μυϊκή Δυστροφία Duchenne.")
6
Νόσος του Huntington (Huntington Chorea) Αυτή η γενετική νόσος πρωτοπεριγράφτηκε απο τον Huntington το 1872, σ' έναν Αμερικανό αγγλικής καταγωγής. Στην πραγματικότητα το γονίδιο έχει εξαπλωθεί από την Β.Δ. Ευρώπη σ' όλο τον κόσμο. Η νόσος του Huntington οφείλεται σ' ένα αυτοσωμικό κυρίαρχο γονίδιο, το οποίο βρίσκεται στο μικρό βραχίονα του 4ου (αυτοσωμικού) χρωμοσώματος. Πολύ λίγοι ασθενείς είναι νέα μεταλλάγματα, αφού σχεδόν όλοι οι ασθενείς έχουν άρρωστο γονιό- το οικογενειακό ιστορικό χρησιμοποιείται συχνά σαν ένα από τα διαγνωστικά κριτήρια. Η παρουσία του γονιδίου της HC προξενεί νοητική υστέρηση και μη φυσιολογικές (σπαστικές) κινήσεις των άκρων χωρίς συνειδητό έλεγχο (chorea). Αρχίζει μεταξύ των 20-40 χρόνων και εξελίσσεται σε αδυναμία, κατάρρευση και άνοια και τελικά επέρχεται ο θάνατος, ο οποίος είναι αποτέλεσμα δευτερογενών λοιμώξεων, καρδιακής ανεπάρκειας ή πνευμονίας. Έχει χαρακτηρισθεί σαν "η πιο δαιμονική από τις αρρώστιες" και στο παρελθόν πολλές ιστορίες για κατάλειψη από δαίμονες και μαγεία πρέπει να έχουν προέλθει από την συμπεριφορά ασθενών με νόσο του Huntington. Δεν υπάρχει θεραπεία, αλλά υπάρχει προγεννητικό διαγνωστικό test.
 χρωμοσώματος. Πολύ λίγοι ασθενείς είναι νέα μεταλλάγματα, αφού σχεδόν όλοι οι ασθενείς έχουν άρρωστο γονιό- το οικογενειακό ιστορικό χρησιμοποιείται συχνά σαν ένα από τα διαγνωστικά κριτήρια. Η παρουσία του γονιδίου της HC προξενεί νοητική υστέρηση και μη φυσιολογικές (σπαστικές) κινήσεις των άκρων χωρίς συνειδητό έλεγχο (chorea). Αρχίζει μεταξύ των χρόνων και εξελίσσεται σε αδυναμία, κατάρρευση και άνοια και τελικά επέρχεται ο θάνατος, ο οποίος είναι αποτέλεσμα δευτερογενών λοιμώξεων, καρδιακής ανεπάρκειας ή πνευμονίας. Έχει χαρακτηρισθεί σαν η πιο δαιμονική από τις αρρώστιες και στο παρελθόν πολλές ιστορίες για κατάλειψη από δαίμονες και μαγεία πρέπει να έχουν προέλθει από την συμπεριφορά ασθενών με νόσο του Huntington. Δεν υπάρχει θεραπεία, αλλά υπάρχει προγεννητικό διαγνωστικό test..")
7
Νόσος του Huntington (Huntington Chorea)
")
8
Primordial Dwarfism What Causes Primordial Dwarfism? Medical researchers are still in the dark regarding the exact causes of Primordial Dwarfism. The condition is not believed to arise due to any deficiency of nutrition or growth hormone. The disorder is supposed to arise primarily due to genetic factors. It is thought to be an acquired hereditary condition that develops when an embryo receives a copy of defective gene from each parent. The condition may not be apparent in parents. However, they might have the defective gene in a recessive fashion.

11
Σύνδρομο Ellis-van Creveld Μια σπάνια μορφή νανισμού το σύνδρομο Ellis-van Creveld, χαρακτηρίζεται και από πολυδακτυλία και νανισμό, αλλά και ανωμαλίες των νυχιών και των δοντιών, και σε περίπου τα μισά από τα άτομα, μία οπή ανάμεσα ατον κόλπον και την κοιλία της καρδιάς. Το σύνδρομο είναι κοινό στους Amish εξαιτίας του «Φαινομένου του Ιδρυτή»: Όταν μια μικρή ομάδα πληθυσμού μεταναστεύει σε νέα περιοχή, τα γονίδια των «Ιδρυτών» στη νέα κοινωνία είναι δισανάλογα συχνά.

12
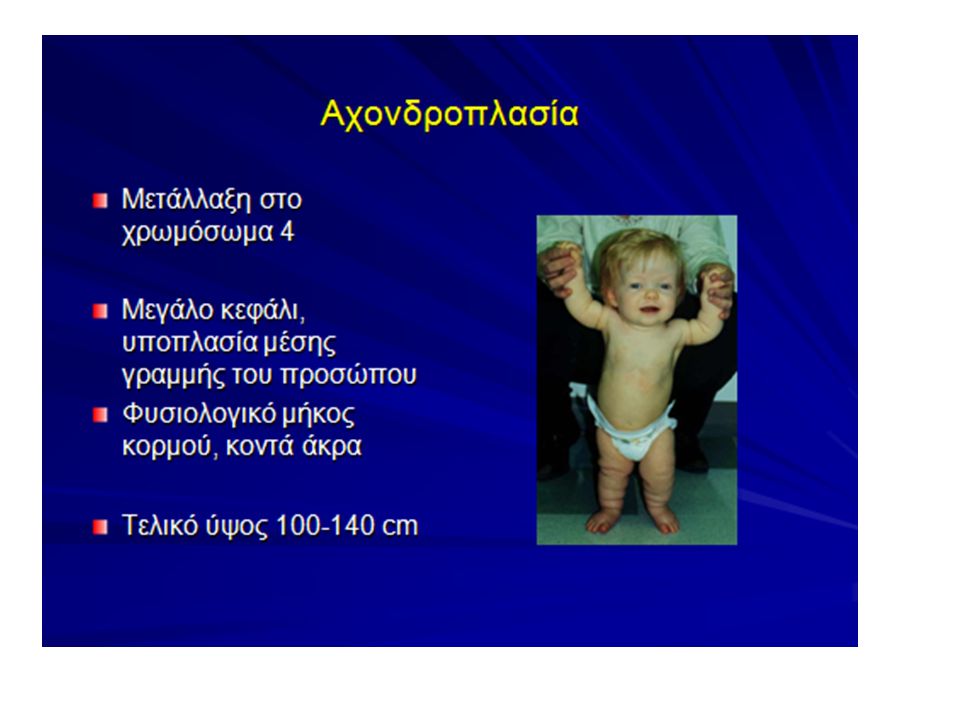
Γενετικό νόσημαΕίδος γονιδίουΣυμπτώματα ΡετινοβλάστωμαΕπικρατές αυτοσωμικό (Ε.Α.) Νεοπλασία στο μάτι. Παιδική αμαυρωτική ιδιοτία Υπολειπόμενο αυτοσωμικό (Υ.Α.) (Κοινό στις Σκανδι- ναβικές χώρες) Τύφλωση, παράλυση, πνευματική καθυστέρηση, θάνατος περίπου στο 6ο έτος. Βρεφική αμαυρωτική ιδιοτία (Ασθένεια Tay- Sacchs) Y.A.Ίδια συμπτώματα. Θάνατος γύρω στα 2 έτη. Αχονδροπλαστικός νανισμός Σύνδρομο Ellis-van Creveld Ε.Α. Y.A Κοντά άκρα. + πολυδακτυλία ΑιμοφιλίαΦυλοσύνδετο υπολειπόμενο (Φ.Υ.) Αργή πήξη του αίματος. Μυϊκή δυστροφίαΦ.Υ.Δυσπλασία μυών. ΑνιριδισμόςΕ.Α.Απουσία ίριδας από τα μάτια. ΑλφισμόςΥ.Α.Απουσία μελανίνης. Κυστική ίνωσηΥ.Α.Προσβάλλει 1/20.000 παιδιά. Δημιουργία βλέννας. Φράξιμο πνευμόνων και πεπτικού.
 (Κοινό στις Σκανδι- ναβικές χώρες) Τύφλωση, παράλυση, πνευματική καθυστέρηση, θάνατος περίπου στο 6ο έτος. Βρεφική αμαυρωτική ιδιοτία (Ασθένεια Tay- Sacchs) Y.A.Ίδια συμπτώματα. Θάνατος γύρω στα 2 έτη. Αχονδροπλαστικός νανισμός Σύνδρομο Ellis-van Creveld Ε.Α. Y.A Κοντά άκρα. + πολυδακτυλία ΑιμοφιλίαΦυλοσύνδετο υπολειπόμενο (Φ.Υ.) Αργή πήξη του αίματος. Μυϊκή δυστροφίαΦ.Υ.Δυσπλασία μυών. ΑνιριδισμόςΕ.Α.Απουσία ίριδας από τα μάτια. ΑλφισμόςΥ.Α.Απουσία μελανίνης. Κυστική ίνωσηΥ.Α.Προσβάλλει 1/ παιδιά. Δημιουργία βλέννας. Φράξιμο πνευμόνων και πεπτικού..")
13
Μεσογειακή αναιμία: Είναι η πιο συνηθισμένη περίπτωση για την Ελλάδα και οφείλεται στο γεγονός ότι συντίθεται σε μικρότερη ποσότητα ή δε συντίθεται καθόλου β-αλυσίδα στην αιμοσφαιρίνη. Λιγότερη β-αλυσίδα συντίθεται όταν το άτομο είναι φορέας (ετεροζυγώτης) του ελαττωματικού γονιδίου (στίγμα). Σ' αυτή την περίπτωση το ολικό ποσό των β- αλυσίδων είναι λιγότερο απ' το φυσιολογικό, αλλά παρόλα αυτά η αιμοσφαιρίνη που παράγουν τα άτομα είναι αρκετή για μια φυσιολογική ζωή. Αντίθετα, στην ομόζυγη κατάσταση δεν συντίθεται καθόλου αλυσίδα β στην αντίστοιχη αιμοσφαιρίνη, με αποτέλεσμα το άτομο να εμφανίζει τη βαρύτερη εικόνα της β-Μεσογειακής αναιμίας (νόσος Cooley, από το όνομα του Αμερικανού παιδίατρου που πρώτος την περιέγραψε το 1925).
 του ελαττωματικού γονιδίου (στίγμα). Σ αυτή την περίπτωση το ολικό ποσό των β- αλυσίδων είναι λιγότερο απ το φυσιολογικό, αλλά παρόλα αυτά η αιμοσφαιρίνη που παράγουν τα άτομα είναι αρκετή για μια φυσιολογική ζωή. Αντίθετα, στην ομόζυγη κατάσταση δεν συντίθεται καθόλου αλυσίδα β στην αντίστοιχη αιμοσφαιρίνη, με αποτέλεσμα το άτομο να εμφανίζει τη βαρύτερη εικόνα της β-Μεσογειακής αναιμίας (νόσος Cooley, από το όνομα του Αμερικανού παιδίατρου που πρώτος την περιέγραψε το 1925)..")
14
- Tα γονίδια που αποτελούν ένα ζεύγος μπορεί να είναι όμοια ή ανόμοια. Στην πρώτη περίπτωση μιλάμε για ομοζυγωτία και για ομόζυγα άτομα ενώ στη δεύτερη για ετεροζυγωτία και ετερόζυγα άτομα. Ο φαινοτυπικός χαρακτήρας που αναπτύσσεται από ένα ετερόζυγο άτομο ονομάζεται επικρατής και ο χαρακτήρας που δεν αναπτύσσεται, μολονότι υπάρχει το γονίδιο που συνδέεται μ' αυτόν, ονομάζεται υπολειπόμενος. Για παράδειγμα, το γονίδιο που προξενεί τον αλφισμό στον άνθρωπο (α) είναι υπολειπόμενο ως προς το κανονικό γονίδιο (Α). Ένα ετεροζυγωτικό άτομο (Αα) θα φαίνεται κανονικό ενώ για να είναι κάποιος αλφικός θα πρέπει να είναι ομοζυγωτικό για το υπολειπόμενο γονίδιο (αα).
 είναι υπολειπόμενο ως προς το κανονικό γονίδιο (Α). Ένα ετεροζυγωτικό άτομο (Αα) θα φαίνεται κανονικό ενώ για να είναι κάποιος αλφικός θα πρέπει να είναι ομοζυγωτικό για το υπολειπόμενο γονίδιο (αα)..")
15
Μεσογειακή αναιμία: Είναι η πιο συνηθισμένη περίπτωση για την Ελλάδα και οφείλεται στο γεγονός ότι συντίθεται σε μικρότερη ποσότητα ή δε συντίθεται καθόλου β-αλυσίδα στην αιμοσφαιρίνη. Λιγότερη β-αλυσίδα συντίθεται όταν το άτομο είναι φορέας (ετεροζυγώτης) του ελαττωματικού γονιδίου (στίγμα). Σ' αυτή την περίπτωση το ολικό ποσό των β- αλυσίδων είναι λιγότερο απ' το φυσιολογικό, αλλά παρόλα αυτά η αιμοσφαιρίνη που παράγουν τα άτομα είναι αρκετή για μια φυσιολογική ζωή. Αντίθετα, στην ομόζυγη κατάσταση δεν συντίθεται καθόλου αλυσίδα β στην αντίστοιχη αιμοσφαιρίνη, με αποτέλεσμα το άτομο να εμφανίζει τη βαρύτερη εικόνα της β-Μεσογειακής αναιμίας (νόσος Cooley, από το όνομα του Αμερικανού παιδίατρου που πρώτος την περιέγραψε το 1925).
 του ελαττωματικού γονιδίου (στίγμα). Σ αυτή την περίπτωση το ολικό ποσό των β- αλυσίδων είναι λιγότερο απ το φυσιολογικό, αλλά παρόλα αυτά η αιμοσφαιρίνη που παράγουν τα άτομα είναι αρκετή για μια φυσιολογική ζωή. Αντίθετα, στην ομόζυγη κατάσταση δεν συντίθεται καθόλου αλυσίδα β στην αντίστοιχη αιμοσφαιρίνη, με αποτέλεσμα το άτομο να εμφανίζει τη βαρύτερη εικόνα της β-Μεσογειακής αναιμίας (νόσος Cooley, από το όνομα του Αμερικανού παιδίατρου που πρώτος την περιέγραψε το 1925)..")
17
Οι αριθμοί αυτοί υποδηλώνουν ότι κάθε χρόνο στον ελληνικό χώρο γεννιούνται 100-150 παιδιά με ομόζυγη μορφή Μεσογειακής αναιμίας και άλλων συνδυασμών της. Πραγματικά, μέχρι τελευταία, ο αριθμός των γεννήσεων παιδιών με Μεσογειακή αναιμία έφτανε ή ξεπερνούσε τα 250 ετησίως. Σήμερα, με τη βελτίωση των προγραμμάτων πρόληψης, ο αριθμός αυτός έχει περιοριστεί στα 10-15 ετησίως. Επιπλέον, ενώ οι πάσχοντες από τη νόσο στις αρχές της δεκαετίας του '60 μόλις που κατάφερναν να φθάσουν τα 20 χρόνια ζωής, σήμερα, ιδιαίτερα μετά την ανακάλυψη της ουσίας (δεσφεριοξαμίνη) που βοηθάει στην αποσηδήρωση των πασχόντων, τα άτομα με Μ.Α. μπορούν να έχουν μια ζωή που σχεδόν να μη διαφέρει από τη ζωή των άλλων συνανθρώπων τους.
 που βοηθάει στην αποσηδήρωση των πασχόντων, τα άτομα με Μ.Α. μπορούν να έχουν μια ζωή που σχεδόν να μη διαφέρει από τη ζωή των άλλων συνανθρώπων τους..")
18
Μεσογειακή αναιμία: Είναι η πιο συνηθισμένη περίπτωση για την Ελλάδα και οφείλεται στο γεγονός ότι συντίθεται σε μικρότερη ποσότητα ή δε συντίθεται καθόλου β-αλυσίδα στην αιμοσφαιρίνη. Λιγότερη β-αλυσίδα συντίθεται όταν το άτομο είναι φορέας (ετεροζυγώτης) του ελαττωματικού γονιδίου (στίγμα). Σ' αυτή την περίπτωση το ολικό ποσό των β- αλυσίδων είναι λιγότερο απ' το φυσιολογικό, αλλά παρόλα αυτά η αιμοσφαιρίνη που παράγουν τα άτομα είναι αρκετή για μια φυσιολογική ζωή. Αντίθετα, στην ομόζυγη κατάσταση δεν συντίθεται καθόλου αλυσίδα β στην αντίστοιχη αιμοσφαιρίνη, με αποτέλεσμα το άτομο να εμφανίζει τη βαρύτερη εικόνα της β-Μεσογειακής αναιμίας (νόσος Cooley, από το όνομα του Αμερικανού παιδίατρου που πρώτος την περιέγραψε το 1925). Το μέτρο αυτό που στην Ελλάδα δεν έχει εφαρμοστεί (έχει εφαρμόσει όμως η Εκκλησία της Κύπρου) αποδεικνύει περίτρανα τη σημαντικοτητά του κρίνοντας από τα αποτελέσματα που έχει η Κύπρος στις γεννήσεις παιδιών με Μεσογειακή Αναιμία (ποσοστό γεννήσεων τα τελευταία χρόνια=0%).
 του ελαττωματικού γονιδίου (στίγμα). Σ αυτή την περίπτωση το ολικό ποσό των β- αλυσίδων είναι λιγότερο απ το φυσιολογικό, αλλά παρόλα αυτά η αιμοσφαιρίνη που παράγουν τα άτομα είναι αρκετή για μια φυσιολογική ζωή. Αντίθετα, στην ομόζυγη κατάσταση δεν συντίθεται καθόλου αλυσίδα β στην αντίστοιχη αιμοσφαιρίνη, με αποτέλεσμα το άτομο να εμφανίζει τη βαρύτερη εικόνα της β-Μεσογειακής αναιμίας (νόσος Cooley, από το όνομα του Αμερικανού παιδίατρου που πρώτος την περιέγραψε το 1925). Το μέτρο αυτό που στην Ελλάδα δεν έχει εφαρμοστεί (έχει εφαρμόσει όμως η Εκκλησία της Κύπρου) αποδεικνύει περίτρανα τη σημαντικοτητά του κρίνοντας από τα αποτελέσματα που έχει η Κύπρος στις γεννήσεις παιδιών με Μεσογειακή Αναιμία (ποσοστό γεννήσεων τα τελευταία χρόνια=0%)..")
19
Ο ανθρώπινος οργανισμός έχει περιορισμένη δυνατότητα απέκκρισης σιδήρου. Το γεγονός αυτό επιβαρύνει σημαντικά τον οργανισμό των πασχόντων από μεσογειακή αναιμία, καθώς η μετάγγιση 250 ml συμπυκνωμένων ερυθρών αιμοσφαιρίων προσθέτει στον οργανισμό τους περίπου 250 mg σιδήρου. Οι επιπλοκές από την υπερφόρτωση με σίδηρο είναι πολλές, με πιο σοβαρές την κίρρωση του ήπατος, τον ηπατοκυτταρικό καρκίνο, την καρδιακή κάμψη, το σακχαρώδη διαβήτη και τη δυσλειτουργία της υπόφυσης. Η δεσφεριοξαμίνη, αυξάνει την απέκκριση του σιδήρου μέσω της ούρησης. Όμως, χορηγείται είτε ενδοφλέβια είτε με συνεχή υποδόρια έκχυση, γεγονός που σημαίνει ότι οι ασθενείς θα πρέπει να υποβάλλονται σε περίπου 12 ώρες συνεχούς υποδόριας έκχυσης 4-6 φορές την εβδομάδα. Η έκχυση γίνεται με ειδικές αντλίες που είναι άβολες, με αποτέλεσμα πολλοί ασθενείς να μην ακολουθούν τη θεραπεία. Αν στο γεγονός αυτό προστεθεί και ο κίνδυνος σοβαρών παρενεργειών καθώς και το υψηλό κόστος της θεραπείας, γίνεται κατανοητό γιατί η φαρμακευτική έρευνα έχει στραφεί στην ανακάλυψη κάποιου φαρμάκου, κατά προτίμηση με τη μορφή χαπιού, που θα έχει λιγότερες παρενέργειες. Ένα τέτοιο φάρμακο που δοκιμάζεται τελευταία με θετικά αποτελέσματα είναι η δεφεριπρόνη. Δεσφεροξαμίνη και Δεφεριπρόνη

20
Απαντά συνολικά σε 100 εκατομμύρια ανθρώπους σ' όλο τον κόσμο και ιδιαίτερα σε περιοχές όπου ενδημούσε η ελονοσία. Στις ΗΠΑ απαντά σε ένα ποσοστό 10% των νέγρων. Πρόκειται για φυλοσύνδετο συνεπικρατή χαρακτήρα. Κάτω από κανονικές συνθήκες, οι φορείς αυτού του γονιδίου είναι κανονικά άτομα. Όταν όμως τους χορηγηθεί κάποιο ανθελονοσιακό φάρμακο ή σουλφοναμίδη ή ασπιρίνη δημιουργούν αναιμία. Tα ίδια συμπτώματα μπορεί να δημιουργηθούν από την επαφή με ναφθαλίνη ή τη λήψη χλωρών κουκιών (Κυαμισμός). Δεν υπάρχει λόγος ανησυχίας για κάποια επιπλοκή στο μέλλον, εκτός των φαρμάκων που μας απαγορεύεται να χορηγηθούν, μιας και υπάρχουν εναλλακτικά που δεν έχουν παρενέργειες. Καλό είναι επίσης να βλέπουμε και τις αντενδείξεις κάθε φαρμάκου πριν τη λήψη του, ούτως ώστε αν αναφέρεται ως ακατάλληλο για αυτούς που έχουν την έλλειψη, να μην το πάρουμε. Επίσης απαγορεύεται να τρώμε κουκιά-φάβα και να εισπνέουμε ναφθαλίνη. Ανεπάρκεια G6ΡD
. Δεν υπάρχει λόγος ανησυχίας για κάποια επιπλοκή στο μέλλον, εκτός των φαρμάκων που μας απαγορεύεται να χορηγηθούν, μιας και υπάρχουν εναλλακτικά που δεν έχουν παρενέργειες. Καλό είναι επίσης να βλέπουμε και τις αντενδείξεις κάθε φαρμάκου πριν τη λήψη του, ούτως ώστε αν αναφέρεται ως ακατάλληλο για αυτούς που έχουν την έλλειψη, να μην το πάρουμε. Επίσης απαγορεύεται να τρώμε κουκιά-φάβα και να εισπνέουμε ναφθαλίνη. Ανεπάρκεια G6ΡD.")
27
Tο σύνδρομο Κlinefelter και το σύνδρομο Triplo-Χ είναι δύο σύνδρομα που οφείλονται σε τρισωμίες των χρωμοσωμάτων του φύλου. Έτσι στο σύνδρομο Κlinefelter o γονότυπος είναι ΧΧΥ και τα άτομα που το φέρουν είναι φαινοτυπικά άρρενα που χαρακτηρίζονται από γυναικομαστία και αζωοσπερμία. Αντίστοιχα, στο Triplo-Χ ο γονότυπος είναι ΧΧΧ, ενώ ο φαινότυπος χαρακτηρίζεται από το γεγονός ότι τα άτομα αυτά ανήκουν στο γυναικείο φύλο και χαρακτηρίζονται από ατελή ανάπτυξη των δευτερευόντων χαρακτήρων του φύλου (αυξημένο τρίχωμα, βαριά φωνή κ.λ.π.).
..")
31
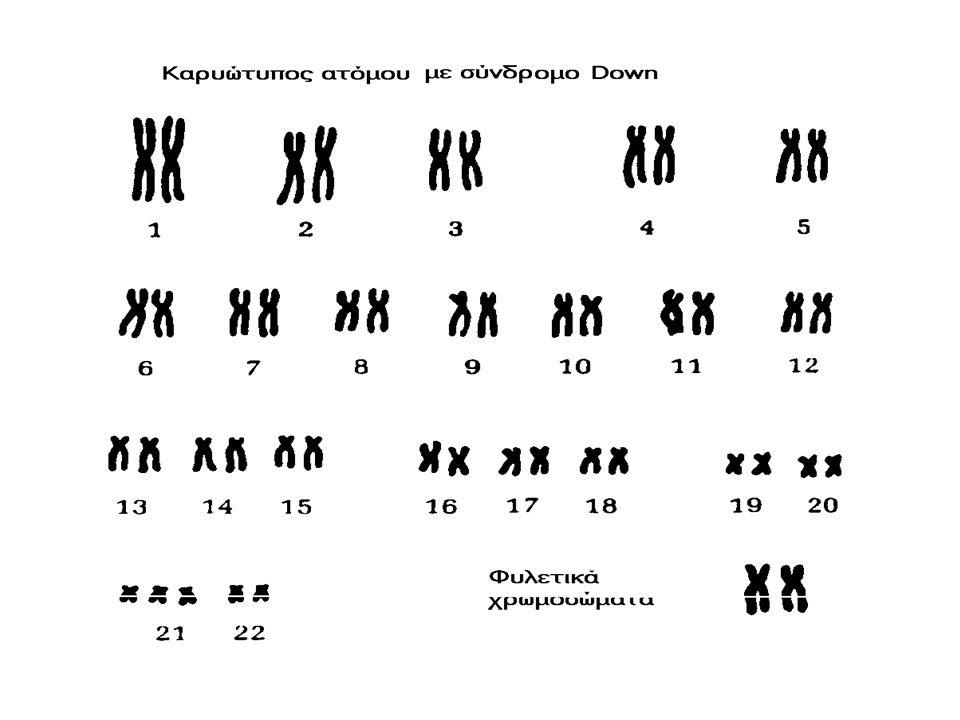
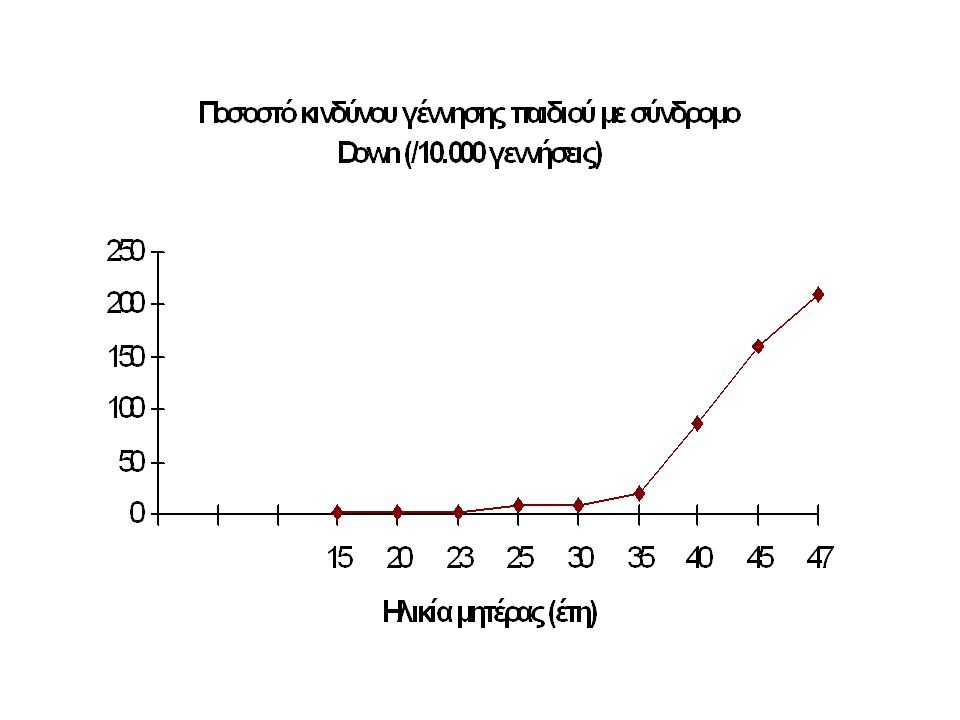
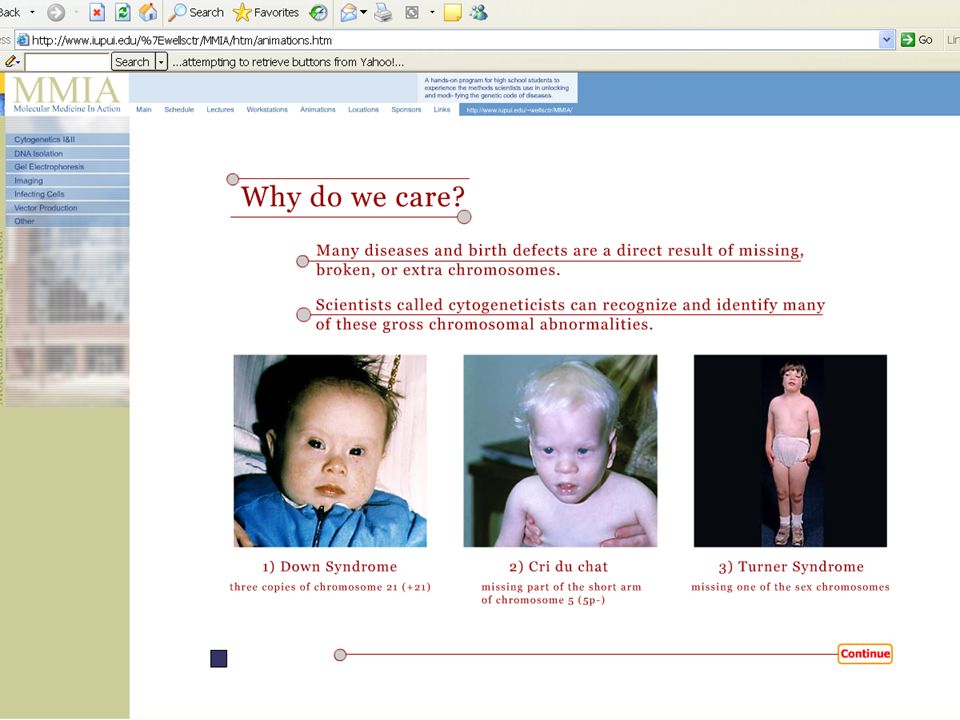
Down Syndrome (Trisomy 21) Down Syndrome is a genetic disorder caused by extra genetic material. It affects over 350,000 people in the United States alone and is the most common (1 in 800 live births) imbalance in the number of autosomes in people. The effects of Down Syndrome vary greatly from person to person but can include mental retardation, eyes that slant upward, and heart defects. People with Down Syndrome have 3 copies of chromosome 21. For this reason, Down Syndrome is also called "Trisomy 21". Where does the extra chromosome come from? In 90% of Trisomy 21 cases, the additional chromosome comes from the mother's egg.
 imbalance in the number of autosomes in people. The effects of Down Syndrome vary greatly from person to person but can include mental retardation, eyes that slant upward, and heart defects. People with Down Syndrome have 3 copies of chromosome 21. For this reason, Down Syndrome is also called Trisomy 21 . Where does the extra chromosome come from. In 90% of Trisomy 21 cases, the additional chromosome comes from the mother s egg..")
32
Σύνδρομο Down Απαντά σε 1/850 βιώσιμες γεννήσεις στις ΗΠΑ. (350.000 άτομα). Στο 90% των περιπτώσεων το επι πλέον χρωμόσωμα προέρχεται από το ωάριο της μητέρας. Συνήθη χαρακτηριστικά: Πιθανή νοητική στέρηση, ΄χαρακτηριστικό σχήμα ματιών, και καρδιακή ανεπάρκεια.
. Στο 90% των περιπτώσεων το επι πλέον χρωμόσωμα προέρχεται από το ωάριο της μητέρας. Συνήθη χαρακτηριστικά: Πιθανή νοητική στέρηση, ΄χαρακτηριστικό σχήμα ματιών, και καρδιακή ανεπάρκεια..")
33
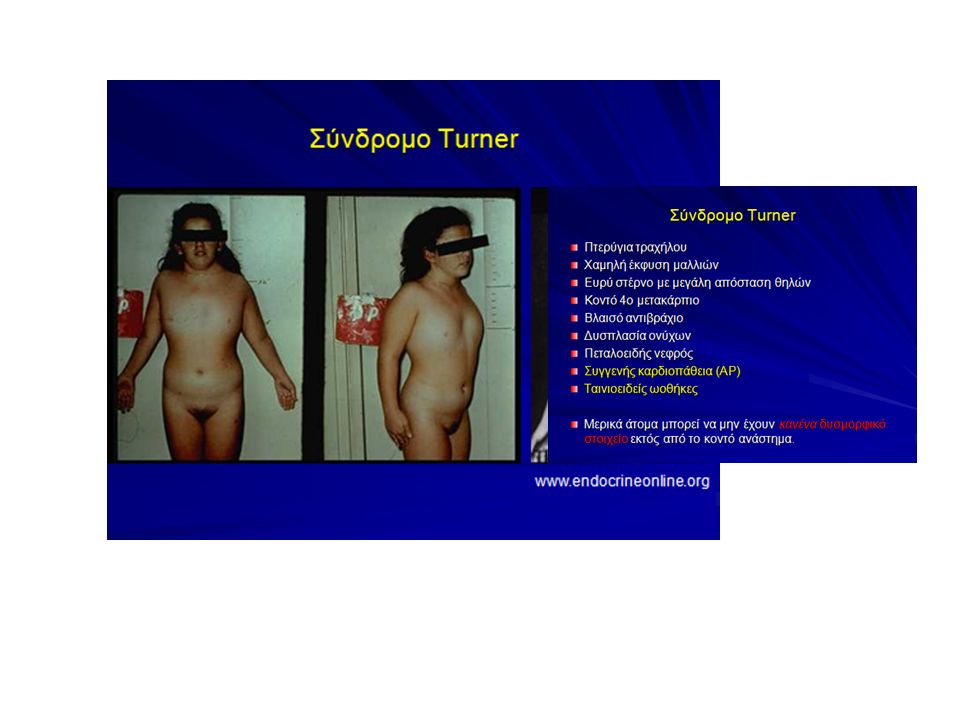
Turner Syndrome

34
ΣΥΝΔΡΟΜΟ TURNER: Αριθμός περιπτώσεων στις ΗΠΑ: 60,000 κορίτσια. 1/2000 έως 1/2,500 γεννήσεις ζώντων κοριτσιών τον χρόνο. ΣΥΜΠΤΩΜΑΤΑ: Κοντό ύψος, ελαφρά απόκλιση του βραχίονα προς τα έξω στο ύψος του αγκώνα, χαμηλή τριχοφυϊα στο πίσω μέρος της κεφαλής. Ύπαρξη ενός μόνο χρωμοσώματος Χ.

36
Σύνδρομο Klinefelter Καρυώτυπος: ΧΧΨ. Χαρακτηριστικά: Συνήθως υψηλόσωμα άτομα, με έλλειψη των δευτερευόντων χαρακτηριστικών του φύλλου: τρίχωμα προσώπου και μασχάλης

37
Cri du chat Syndrome Έλλειψη ενός μέρους του Χρωμοσώματος-5. (Μάλλον πατρική προέλευση). Χαρακτηριστικό κλάμα που οφείλεται σε ανώμαλη ανάπτυξη του λάρυγγα η οποία διορθώνεται σε μερικές εβδομάδες μετά τη γέννα. Χαμηλό βάρος σώματος και πιθανά αναπνευστικά προβλήματα ή χαμηλό αναμενόμενο προσδόκιμο ζωής.
. Χαρακτηριστικό κλάμα που οφείλεται σε ανώμαλη ανάπτυξη του λάρυγγα η οποία διορθώνεται σε μερικές εβδομάδες μετά τη γέννα. Χαμηλό βάρος σώματος και πιθανά αναπνευστικά προβλήματα ή χαμηλό αναμενόμενο προσδόκιμο ζωής..")
38
Reciprocal Translocation: Philadelphia Chromosome Αμοιβαία μετατόπιση τμήματων στα χρωμοσώματα 9 και 22. Τα άτομα αυτά αναπτύσσουν Χρόνοια Μυελογενή Λευκαιμία. Αλλοίωση του γονιδίου abl..

39
Williams Syndrome

40
Σύνδρομο Williams Έλλειψη ενός πολύ μικρού τμήματος του Χρωμοσώματος-7. Έλλειψη του γονιδίου που ελέγχει την πρωτεϊνη Ελαστίνη. Η τελευταία προσδίδει στα αγγεία την ελαστικότητα και την αντοχή. Αγγειακές παθήσεις.

41
Τα αποτελέσματα ποικίλουν: Κάποιες κοπέλες με επιπλέον Χρωμόσωμα Χ δεν παρουσιάζουν σχεδόν κανένα σύμπτωμα. Κάποιες άλλες παρουσιάζουν Δυσκολίες στη μάθηση Καθυστέρηση στην ανάπτυξη κάποια προβλήματα συμπεριφοράς. Κάποιες από τις κοπέλλες παρουσιάζουν μεγαλύτερο ύψος από το κανονικό, ενώ άλλες κινητικά προβλήματα. Τα περισσότερα άτομα αντιδρούν θετικά στη λογοθεραπεία. Η σεξουαλική ανάπτυξη είναι κανονική, οι γυναίκες ΧΧΧ είναι γόνιμες, με μια μικρή αυξημένη πιθανότητα γέννησης παιδιού με αλλαγές στα φυλετικά χρωμοσώματα. ΤριπλοΧ (XXX)
.")
Παρόμοιες παρουσιάσεις


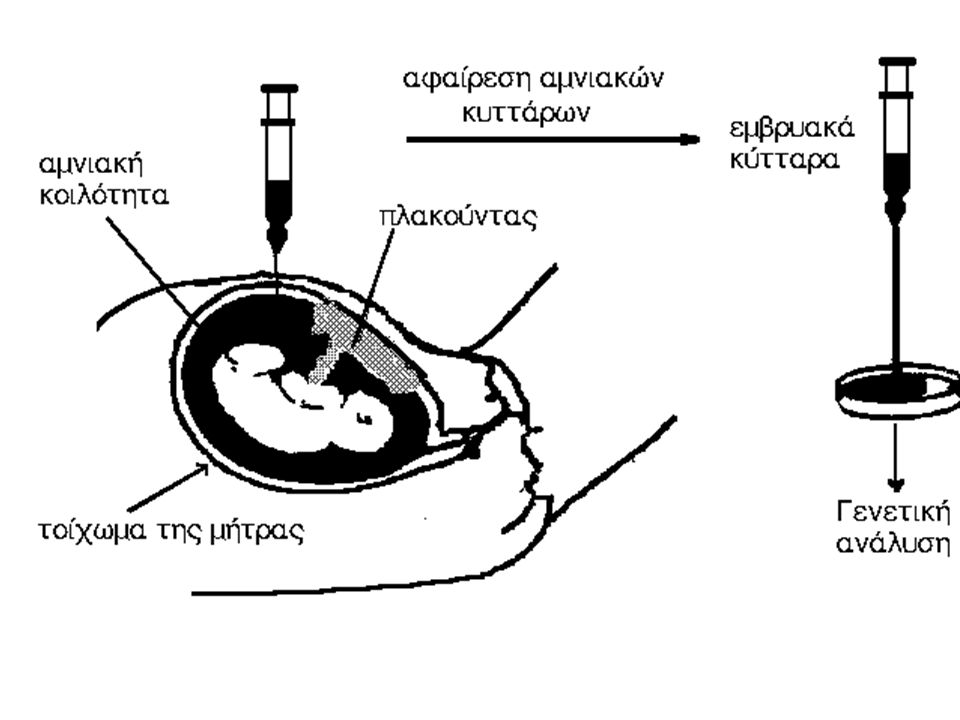


 είναι η ανώμαλη ανάπτυξη κύτταρων με αποτέλεσμα τη δημιουργία όγκων σε διάφορα σημεία του σώματος. Προέλευση της.>")









>")



>")



