Κατέβασμα παρουσίασης
Η παρουσίαση φορτώνεται. Παρακαλείστε να περιμένετε

9
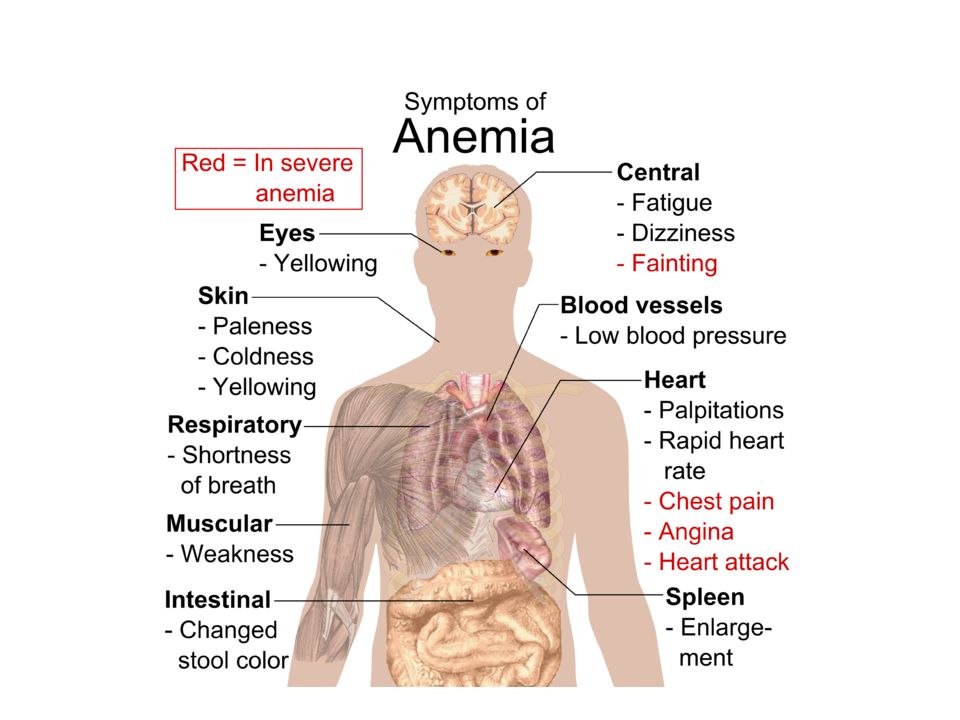
Anemia leads to two symptom complexes;
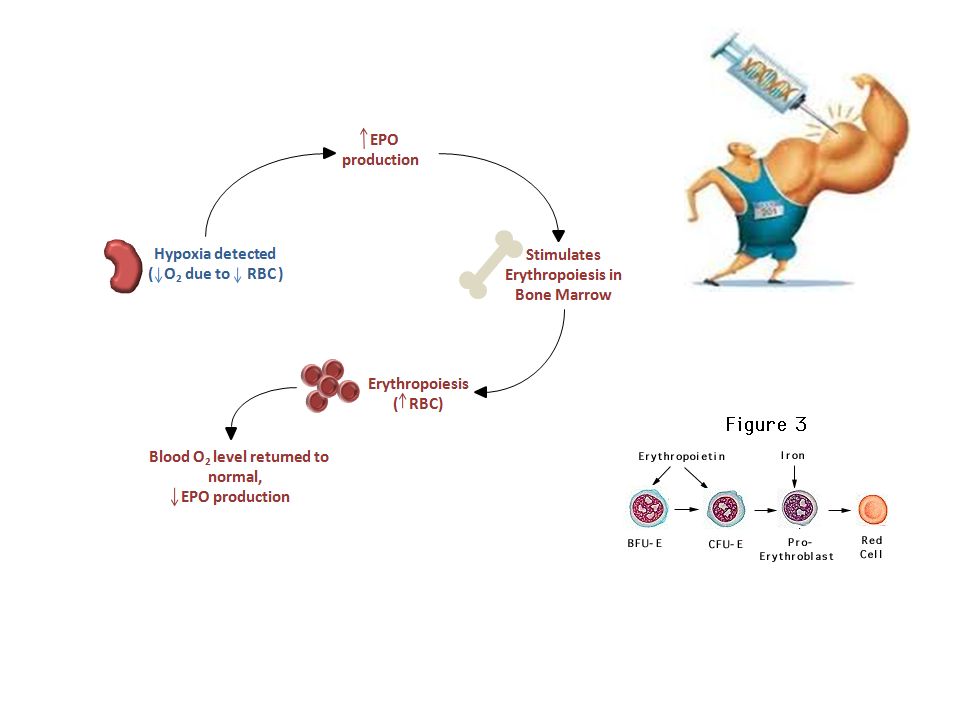
Tissue hypoxia Fatigue,dyspnea on exertion etc Compensatory attempts Tachycardia,hyperventilation etc The most pronounced effects and symptoms derive from skeletal muscles, heart,and central nervous system (due to their greater oxygen demand and compensatory actions). What is the mechanism underlying compensatory mechanisms ? 9
. What is the mechanism underlying compensatory mechanisms 9.")
10
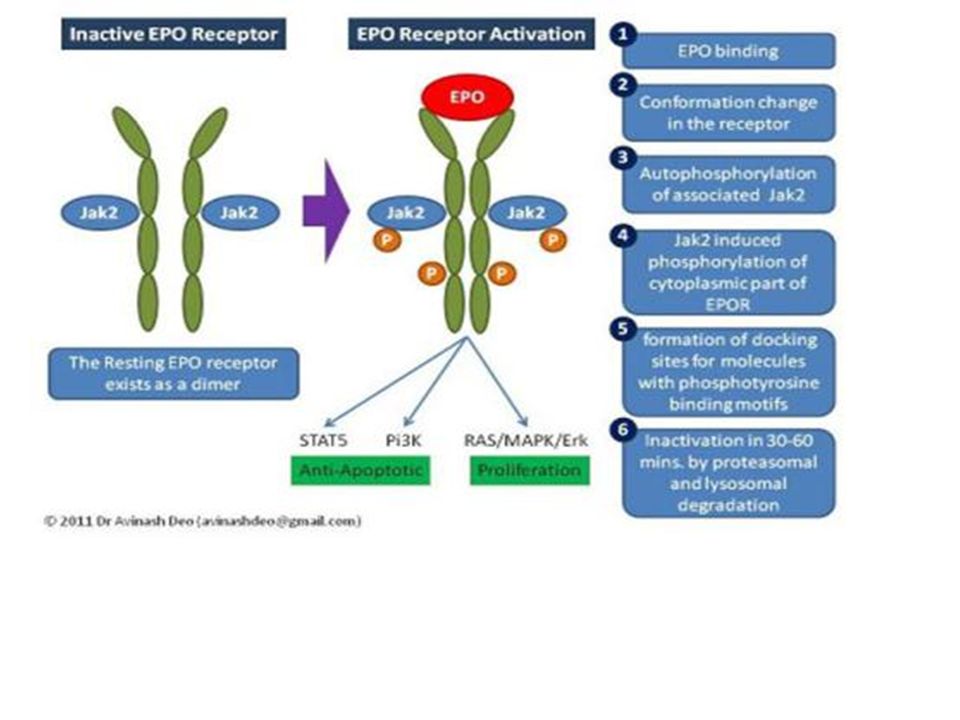
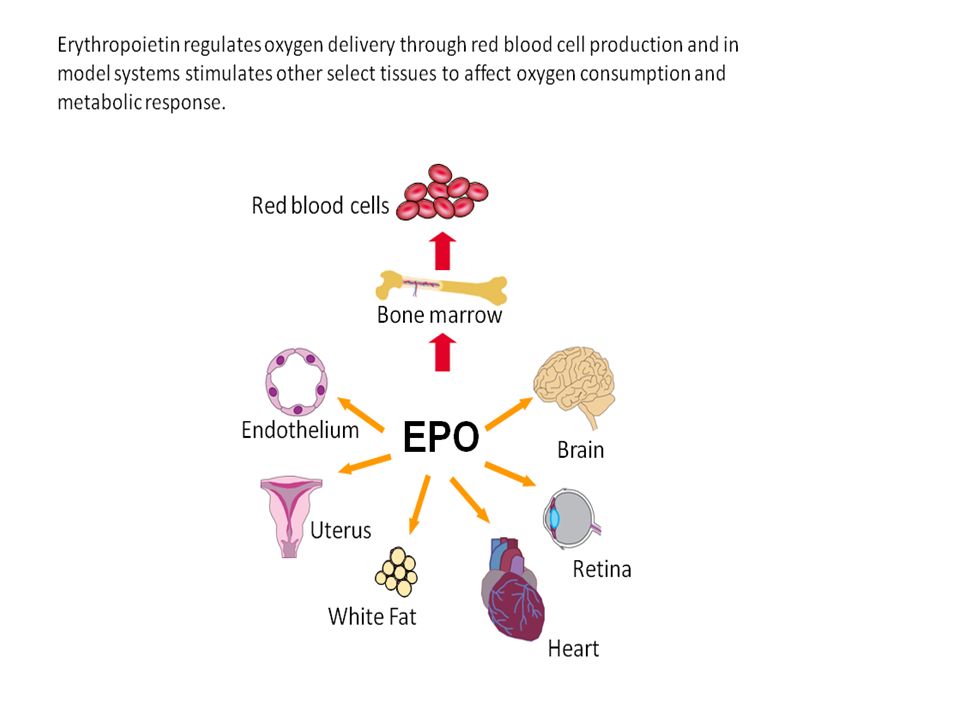
Hypoxia-Inducible Transcription Factor 1
A DNA binding protein Regulated by the O2 tension Regulates genes that promote cell survival under hypoxic conditions Up-reg. EPO gene Up-reg.Glycolytic enzyme genes Up-reg. Angiogenesis Respiratory control Energy metabolism 10

11
HIF-1 Erythropoiesis Angiogenesis and vascular tone EPO
EPO producing cells Vasc. endothelium Muscle heart liver kidney Energy metabolism Decreased O2 consumption HIF-1 All cells Carotid body Glomus cells Iron metabolism Respiration 11

12
Compensating mechanisms in anemia:
The release of oxygen to the tissues is increased (reduced oxygen affinity of Hb) 12
 12.")
13
Compensating mechanisms in anemia
The rate of blood circulation and cardiac output increases. An increase in plasma volume maintains total blood volume in normal or near normal ranges. Redistribution of blood flow. 13

15
Classification of anemia
Morphologic Normocytic: MCV= fL Macrocytic: MCV > 100 fL Microcytic : MCV < 80 fL Pathogenic (underlying mechanism) Blood loss (bleeding) Decreased RBC production Increased RBC destruction/pooling 15
 Blood loss (bleeding) Decreased RBC production. Increased RBC destruction/pooling. 15.")
16
Normocytic Anemias Acute post-hemorrhagic anemia
Hemolytic anemia (except thalassemia and some other Hb disorders) Aplastic anemia Pure red cell aplasia Bone marrow infiltration Endocrin diseases Renal failure Liver disease Chronic disease anemia Protein malnutrition Hypovitaminosis C 16
 Aplastic anemia. Pure red cell aplasia. Bone marrow infiltration. Endocrin diseases. Renal failure. Liver disease. Chronic disease anemia. Protein malnutrition. Hypovitaminosis C. 16.")
17
Microcytic anemias Iron deficiency anemia Thalassemia
Sideroblastic anemia Lead poisoning Anemia of chronic diseases (some cases) 17
 17.")
18
Non-megaloblastic Macrocytic Anemias
Anemia of acute bleeding Hemolytic anemias Leukemias (esp: acute) Myelodysplastic syndromes Liver disease Aplastic anemia Diseases infiltrative to the bone marrow Alcoholism Hypothyroidism Scurvy 18
 Myelodysplastic syndromes. Liver disease. Aplastic anemia. Diseases infiltrative to the bone marrow. Alcoholism. Hypothyroidism. Scurvy. 18.")
19
Pathogenic classification (Causes of anemia)
Decreased RBC production Decreased Hb production Defective DNA synthesis Stem cell defects Pluripotent stem cell Erythroid stem cell(progenitors) Other less defined reasons Blood loss Anemia due to acute bleeding Increased RBC destruction Relative(increased plasma volume) 19
 Other less defined reasons. Blood loss. Anemia due to acute bleeding. Increased RBC destruction. Relative(increased plasma volume) 19.")
20
Decreased Hb production
Defective DNA synthesis Iron deficiency anemia Thalassemia Sideroblastic anemia Lead poisoning Vit B12 deficiency Folic acid deficiency Other.

21
Pluripotent stem cell defects
Aplastic anemia Leukemia or myelodysplastic syndromes Defective erythroid stem cell Pure red cell aplasia Anemia of chronic renal failure Endocrin disease anemia Congenital dyserythropoetic anemias ******Anemia of chronic diseases Bone marrow infiltration Anemia due to nutritional defects 21

22
Lab. investigation of anemia(1)
WBC count and differential Platelet count and morphology ESR Biochemistry, special tests and others Bone marrow exam.(only when indicated) 22
 22.")
23
Lab. investigation of anemia(2)
Serum values of Iron TIBC Ferritin Bilirubins Proteins / electrophoresis LDH Vit B12 and /or Folic acid (None of these tests are routine screening tests) 23
 23.")
24
Lab. Investigation of Anemia(3)
Red cell enzymes Hb F,A2,Hb electrophoresis Coombs tests Liver, renal, endocrin functional tests Urinalysis Hemosiderin Occult GIS bleeding / parasites etc (tests should be chosen individually-do not order routinly ) 24
 24.")
25
Ανδρες 40-54% γυναίκες 37-49%

26
Ανδρες g/dl Γυναίκες g/dl

27
MCV μm3 MCH picograms/cell MCHC g/dl

28
Definition of anemia Hb level of a patient which is below the normal ranges of that age and sex. For adults: WHO criteria define anemia as hemoglobin level lower than 12 g/dL in women and 13 g/dL in men But: The reference values for red cells ,Hb or Hct may difer according to sex/age Race Altitude Socioeconomical changes Study/reference etc 28

29
!!!!! Plasma volume changes have to be considered before determining a diagnosis of anemia . Volume contraction:Underestimation of anemia Volume overload: Underestimation of Hb level 29

30
Volume changes/acute bleeding and anemia
1 2 3 4 5 b a Increased plasma volume Hct: Low Dehydration Hct:Increased normal Hct (a/b%):Normal Acute blood loss(early) Hct:unchanged Chronic anemia Hct: Low 30
:Normal. Acute blood loss(early) Hct:unchanged. Chronic anemia Hct: Low. 30.")
35
Μεθαιμορραγική αναιμία

36
Ενδειξεις βάσει της απώλειας κυκλοφορούντος όγκου
Αν η απώλεια <15% δε μεταγγίζουμε Αν % προτιμούμε κρυσταλλοειδή, συνθετικά κολλοειδή. Αν % τότε χορηγούμε κρυσταλλοειδή, συνθετικά κολλοειδή + μεταγγίζουμε ερυθρά. Αν >40% τότε κάνουμε αναπλήρωση όγκου + μετάγγιση .

39
Cellular Iron Uptake

40
Steap (six-transmembrane epithelial antigen of the prostate) proteins 1–4 ferrireductases,
Steap3 in erythroid precursors , divalent metal transporter-1 (DMT1) multifunctional poly(RC)-binding proteins (PCBPs
 multifunctional poly(RC)-binding proteins (PCBPs.")
41
Cellular Iron Homeostasis
TFR1

42
Regulation of systemic iron
the “stores” regulator, the “erythroid” regulator

43
The Central Role of Hepcidin

44
hepcidin acts by binding to its receptor, ferroportin, and causing its endocytosis and proteolysis,
decreased iron release from cells to plasma and extracellular fluid. Ferroportin is found at very low concentrations in most cell types much higher amounts in the duodenal enterocytes and splenic macrophages.

45
BMP6 stores sensor BMP HFE b2m holotr TFR2 HJV SMAD 4 hepcidin 260 260
matriptase neogenin Smad1,5,8, SMAD 4 hepcidin 260 260 Kb 260

46
Positive regulators of BMP signaling
GPI-linked hemojuvelin enhance acting as a BMP pathway coreceptor

47
σιδηροπενική World’s most common nutritional deficiency
2% in adult men (≤ 69 years old) 10% in Caucasian, non-Hispanic women 19% in African-American women *Value for 1994 CDC. MMWR. 2002;51:899. 47
 10% in Caucasian, non-Hispanic women. 19% in African-American women. *Value for CDC. MMWR. 2002;51:")
48
Main Causes of Anaemia Others 9% Haemolysis 17.5% Iron Deficiency 29%
Acute Bleeding 17.5% Chronic Disease 27% Beris P, Tobler A. Schweiz Rundsch Med Prax. 1997;86:1684. Reprinted from Lambert JF, et al. In C Beaumont, P Beris, Y Beuzard, C Brugnara, eds. Disorders of iron homeostasis, erythrocytes, erythropoiesis. Forum service editore, Genoa, Italy, 2006 page 73 figure 1, by permission of European School of Haemotology. 48

49
Iron Deficiency—Aetiology
Increased demand for iron and/or haematopoiesis Iron loss Decreased iron intake or absorption Adamson JW. In: Kasper DL, ed. Harrison’s Principles Of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005. 49

50
Iron Deficiency—Increased Demand for Iron and/or Haematopoiesis
Infancy and adolescence1,2 Pregnancy and lactation1,2 Low socioeconomic status and poverty greatly increase the prevalence of iron deficiency in this category of populations3 • In patients receiving erythropoietin therapy (= functional iron deficiency)2 1. Adamson JW. In: Kasper DL, ed. Harrison’s Principles Of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005. Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed CDC. MMWR. 2002;51:899. 50
2. 1. Adamson JW. In: Kasper DL, ed. Harrison’s Principles Of Internal Medicine. 16th ed. New York: McGraw-Hill; Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed CDC. MMWR. 2002;51:")
51
Iron Deficiency—Iron loss
In physiologic conditions Menstruation In pathologic conditions Surgery, delivery Haemoglobinuria,haemoptysis Gastrointestinal tract pathology In therapeutic procedures Phlebotomy In blood donation Adamson JW. In: Kasper DL, ed. Harrison’s Principles Of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005: Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed 51

52
Iron Deficiency—Decreased Iron Intake or Absorption
Vegetarians or malnutrition (low-cost diet)1 Malabsorption syndromes Sprue, UHC, and Crohn’s disease2 After gastric and intestinal surgery3 Intestinal parasitosis (ankylostomiasis)3 Helicobacter pylori infection2 Autoimmune atrophic gastritis2 CDC. MMWR. 1998;47(RR-3);1-36. Annabale B, et al. Am J Med. 2001;111:439. Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed 52
1. Malabsorption syndromes. Sprue, UHC, and Crohn’s disease2. After gastric and intestinal surgery3. Intestinal parasitosis (ankylostomiasis)3. Helicobacter pylori infection2. Autoimmune atrophic gastritis2. CDC. MMWR. 1998;47(RR-3);1-36. Annabale B, et al. Am J Med. 2001;111:439. Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed")
53
Iron Deficiency Clinical Manifestations (II)
Skin and conjuctival pallor Koilonychia Angular cheilosis Burning tongue Glossitis Hair loss (alopecia areata) Top figure accessed from: with permission from Nature Publishing Group. Bottom figure accessed from: Modern Nutrition in Health & Disease. 9th ed. Editors: Shils, Olsen, Shike & Ross. Williams & Williams, pub. 53
 Top figure accessed from: with permission from Nature Publishing Group. Bottom figure accessed from: Modern Nutrition in Health & Disease. 9th ed. Editors: Shils, Olsen, Shike & Ross. Williams & Williams, pub. 53.")
54
Iron Deficiency Diagnosis
Laboratory tests for: Iron depletion in the body Iron-deficient erythropoiesis (functional iron deficiency) Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 54
 Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy;")
55
Diagnosis of Iron Depletion in the Body—Haematology
Peripheral blood smear of a patient with severe iron deficient anaemia. Note the important microcytosis (compare red blood cells with lymphocyte) as well as hypochromia, target cells, and poikilocytosis. Graphic courtesy of Dr. P. Beris. 55
 as well as hypochromia, target cells, and poikilocytosis. Graphic courtesy of Dr. P. Beris. 55.")
56
Diagnosis of Iron Depletion in the Body—Haematology
Hypochromic, microcytic anaemia usually with high platelets Differential diagnosis of microcytosis Iron deficiency Thalassaemia syndromes Haemoglobinopathies (E,C,CS, Lepore…) Anaemia of chronic diseases Familial sideroblastic anaemia Miscellaneous (lead intoxication…) Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed 56
 Anaemia of chronic diseases. Familial sideroblastic anaemia. Miscellaneous (lead intoxication…) Hoffman, ed. Hematology: Basic Principles and Practice, 4th ed")
57
Diagnosis of Iron Depletion in the Body—Clinical Chemistry
Serum iron Transferrin (iron binding capacity) Transferrin saturation These parameters are modified by inflammation and by fasting state. They are thus of limited value. Serum ferritin, soluble transferrin receptors (sTfR) and sTfR/log ferritin are excellent tools for screening iron stores Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 57
 Transferrin saturation. These parameters are modified by inflammation and by fasting state. They are thus of limited value. Serum ferritin, soluble transferrin receptors (sTfR) and sTfR/log ferritin are excellent tools for screening iron stores. Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy;")
58
Serum Levels That Differentiate ACD from IDA
Variable ACD IDA Both Conditions Iron Transferrin To normal Transferrin saturation Ferritin Normal to sTfR Normal sTfR/log ferritin Low (<1) High (>2) Cytokine levels 58
 High (>2) Cytokine levels. 58.")
59
Iron Deficiency—Diagnosis
Bone marrow examination for stainable iron was regarded in the past as the gold standard for diagnosing iron deficiency No longer recommended for routine evaluation High inter- and intra-observer variability in evaluation Discomfort associated with procedure Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 59

60
Iron Deficiency—Diagnosis
Patients with IDA and a high risk of underlying disease (eg, men of all ages and postmenopausal women) should be evaluated endoscopically for occult bleeding1 Video capsule endoscopy (VCE) should be considered in suspected small-bowel malignancy2 S Killip, et al. Am Fam Physician. 2007;75:671. Urbain D, et al. Endoscopy. 2006;38:408. 60
 should be evaluated endoscopically for occult bleeding1. Video capsule endoscopy (VCE) should be considered in suspected small-bowel malignancy2. S Killip, et al. Am Fam Physician. 2007;75:671. Urbain D, et al. Endoscopy. 2006;38:")
61
Screening for Iron Deficiency
The US Preventive Services Task Force recommends screening only for pregnant women There is insufficient evidence to support routine screening in other asymptomatic persons S Killip, et al. Am Fam Physician. 2007;75:671. 61

62
Iron-Deficient Erythropoiesis (Functional Iron Deficiency)—Diagnosis
Normal or increased ferritin Laboratory signs of iron-deficient erythropoiesis Serum iron <60 μg/dL Transferrin saturation <20% Hypochromic RBC >5% Reticulocyte Hb content (CHr) <29 pg Soluble transferrin receptor > 7 mg/L Beguin Y, et al. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 62
 <29 pg. Soluble transferrin receptor > 7 mg/L. Beguin Y, et al. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy;")
63
Main Conditions Characterized by Functional Iron Deficiency
EPO-stimulated red cell production (anaemia of chronic kidney disease) Insufficient mobilization of iron from macrophages (anaemia in rheumatoid arthritis and in cancer) Beguin Y, et al. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 63
 Insufficient mobilization of iron from macrophages (anaemia in rheumatoid arthritis and in cancer) Beguin Y, et al. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy;")
64
Refractory Iron Deficiency Anaemia
, Helicobacter pylori coeliac disease Autoimmune atrophic gastritis or atrophic body gastritis Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 64

65
Recommendations for the Diagnostic Work-Up of Refractory IDA
Screening for coeliac disease, autoimmune type A atrophic gastritis and for H. pylori should be performed in the following populations Males and postmenopausal females with IDA and negative endoscopic and radiologic studies Fertile females and children/adolescents refractory to oral iron treatment Hershko C. In: Beaumont C, et al, eds. Disorders of Iron Homeostasis, Erythrocytes, Erythropoiesis. Forum service editore: Genoa, Italy; 2006. 65

66
ΑΙΜΟΛΥΤΙΚΕΣ Για την επιβεβαίωση χρειαζόμαστε - LDH – υψηλή
- Εμμεση χολερυθρίνη υψηλή - Απτοσφαιρίνη χαμηλή Επιβεβαίωση διάγνωσης Αιμολυτική αναιμία

67
ΔΔ αιμολυτικής αναιμίας:
- Εσωτερικό του ερυθροκυττάρου (αιμοσφαιρινοπάθεια, ενζυμοπάθεια) Μεμβράνη του ερυθροκυττάρου Εξωτερικό του ερυθροκυττάρου (υπερσπληνισμός, μηχανική αιμόλυση, άνοση αιμόλυση, τοξικοί/ φλεγμονώδεις παράγοντες)
 Μεμβράνη του ερυθροκυττάρου. Εξωτερικό του ερυθροκυττάρου (υπερσπληνισμός, μηχανική αιμόλυση, άνοση αιμόλυση, τοξικοί/ φλεγμονώδεις παράγοντες)")
68
A Very Simple Classification of Hemolytic Anemias
Intracorpuscular 1- Abnormalities of RBC interior a. Enzyme defects b. Hemoglobinopathies & Thalassemia M 2-RBC membrane abnormalities a. Hereditary spherocytosis, elliptocytosis etc b. Paroxysmal nocturnal hemoglobinuria c. Spur cell anemia 3- Extrinsic factors a. Hypersplenism b. Antibody : immune hemolysis c. Traumatic & Microangiopathic hemolysis d. Infections , toxins , etc Hereditary Extracorpuscular Acquired 68

69
Περιφερικό επίχρισμα - Ανισοκυττάρωση (high RDW) - Πολυχρωματοφιλία (ΔΕΚ) - Σφαιροκύτταρα? Ελλιπτοκύτταρα? Ακανθοκύτταρα? Σχιστοκύτταρα? Δρεπανοκύτταρα?

71
Ενζυμα

72
Heinz body preparation with Crystal violet
Unstable hemoglobin

75
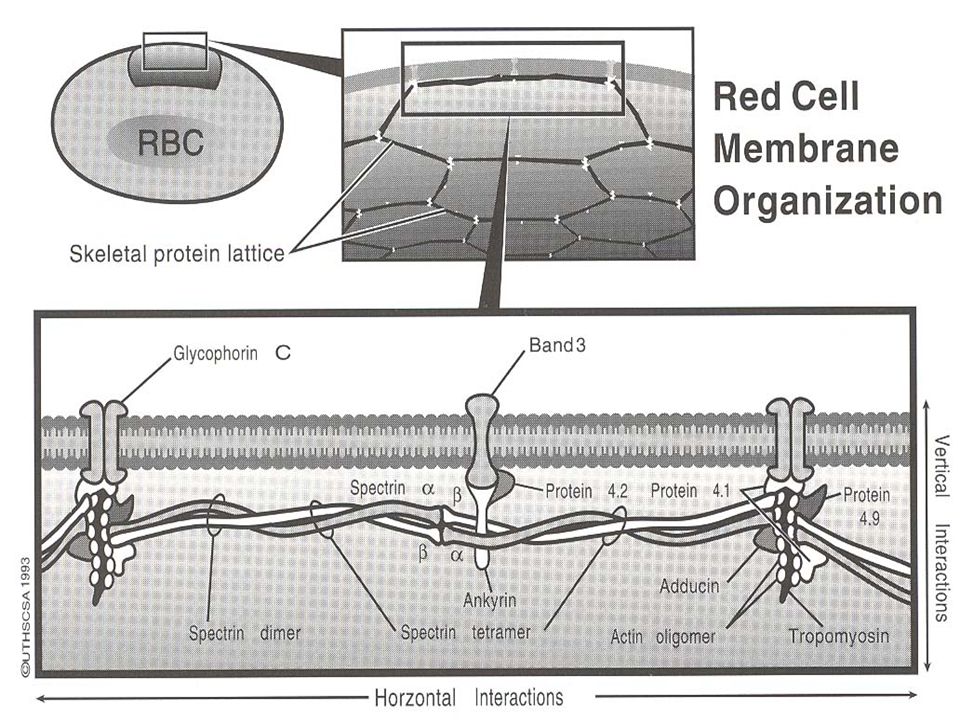
Membrane Defect Spherocytosis Elliptocytosis
PNH (sensitivity to complement lysis -- sugar water test, Ham’s test) Stomatocytosis (possibly Rh null)
 Stomatocytosis (possibly Rh null)")
76
Spherocytes Elliptocytes

77
Stomatocytes

79
Osmotic Fragility

80
What is PNH Mutation? PNH is due to a mutation in a gene in a blood stem cell. The gene is called the PIG-A gene (phosphatidylinositol glycan complementation group A) and is located on the X chromosome. >100 mutations in PIG - A gene known in PNH The gene contains the genetic information for the GPI anchors which link proteins to the cell membrane 80 80
 and is located on the X chromosome. >100 mutations in PIG - A gene known in PNH. The gene contains the genetic information for the GPI anchors which link proteins to the cell membrane")
81
Pathogenesis - The Defect
GPI Anchor PIG - A gene codes for 60 kDa protein glycosyltransferase which effects the first step in the synthesis of the glycolipid GPI anchor (glycosylphosphatidylinositol). Results in clones lacking GPI anchor - in turn, attached proteins PIG - A protein
. Results in clones lacking GPI anchor - in turn, attached proteins. PIG - A protein.")
82
Surface Proteins Missing on PNH Blood Cells
Antigen Expression Pattern Other surface proteins of unknown functions CAMPATH-1 antigen (CDw52) Lymphocytes, monocytes CD B-lymphocytes, Neutrophils, eosinophils p5O Neutrophils GP Platelets GPI Platelets CD55 inhibits the formation or destabilizes complement C3 convertase (C4bC2a) CD59 Protects the membrane from attack by the C5-C9 complex
 Lymphocytes, monocytes. CD24 B-lymphocytes, Neutrophils, eosinophils. p5O-80 Neutrophils. GP500 Platelets. GPI75 Platelets. CD55 inhibits the formation or destabilizes complement C3 convertase (C4bC2a) CD59 Protects the membrane from attack by the C5-C9 complex.")
83
Absence of CD59 Allows Terminal Complement Complex Formation
C5a C8 X X C7 C8 C5b C6 C5b convertase C5 C6 C6 C6 convertase C5 C5b C5b C5b Bb+ C7 CD59 CD59 C7 C7 C3 C3b Shown is a schematic of the terminal complement complex (TCC). The absence of the GPI-anchored complement inhibitor, CD59, allows the formation of C5b-9, which is also known as the membrane attack complex (MAC) or TCC The absence of CD59 increases the sensitivity of PNH RBCs to complement-mediated hemolysis, and accounts for the chronic hemolysis of patients with PNH Adapted from Cellular and Molecular Immunology AK Abbas, AH Litchman and JS Pober, 3rd Edition WB Saunders; Philadelphia. C9 C8 C8 C5b-9 C5b,6,7 C5b-8 Adapted from Cellular and Molecular Immunology AK Abbas, AH Litchman and JS Pober, 3rd Edition WB Saunders; Philadelphia. For Training Purposes Only. Not For Distribution. Alexion Pharmaceuticals, Inc. 83 83
. The absence of the GPI-anchored complement inhibitor, CD59, allows the formation of C5b-9, which is also known as the membrane attack complex (MAC) or TCC. The absence of CD59 increases the sensitivity of PNH RBCs to complement-mediated hemolysis, and accounts for the chronic hemolysis of patients with PNH. Adapted from Cellular and Molecular Immunology AK Abbas, AH Litchman and JS Pober, 3rd Edition WB Saunders; Philadelphia. C9. C8. C8. C5b-9. C5b,6,7. C5b-8. Adapted from Cellular and Molecular Immunology AK Abbas, AH Litchman and JS Pober, 3rd Edition WB Saunders; Philadelphia. For Training Purposes Only. Not For Distribution. Alexion Pharmaceuticals, Inc")
84
84 84

85
PNH Diagnosis by Flow Cytometry
Examples of variable GPI linked CD59 expression on granulocytes on four PNH patients

86
SOLIRIS Blocks Terminal Complement
Antigen-Antibody Complexes Constitutive/ Microorganisms Microorganisms Lectin Classical Alternative SOLIRIS SOLIRIS binds with high affinity to C5 C3 C3a Proximal C3b Terminal complement activity is blocked SOLIRIS binds to C5 and prevents the formation of the terminal complement complex (TCC), also known as the membrane attack complex (MAC) Functions of the TCC including complement-mediated hemolysis are inhibited SOLIRIS does not block proximal complement components, which remain intact Figueroa, et al. Clin Microbiol Rev. 1991;4: Walport. N Engl J Med. 2001;344:1058. SOLIRIS™ (eculizumab) [package insert]. Alexion Pharmaceuticals; 2007. Proximal functions of complement remain intact Weak anaphylatoxin Immune complex and apoptotic body clearance Microbial opsonization C5 C5a Terminal C5b C5b-9 Cause of Hemolysis in PNH Figueroa, et al. Clin Microbiol Rev. 1991;4: Walport. N Engl J Med. 2001;344:1058. SOLIRIS™ (eculizumab) [package insert]. Alexion Pharmaceuticals; 2007. For Training Purposes Only. Not For Distribution. Alexion Pharmaceuticals, Inc. 86 86
, also known as the membrane attack complex (MAC) Functions of the TCC including complement-mediated hemolysis are inhibited. SOLIRIS does not block proximal complement components, which remain intact. Figueroa, et al. Clin Microbiol Rev. 1991;4: Walport. N Engl J Med. 2001;344:1058. SOLIRIS™ (eculizumab) [package insert]. Alexion Pharmaceuticals; Proximal functions of complement remain intact. Weak anaphylatoxin. Immune complex and apoptotic body clearance. Microbial opsonization. C5. C5a. Terminal. C5b. C5b-9. Cause of Hemolysis. in PNH. Figueroa, et al. Clin Microbiol Rev. 1991;4: Walport. N Engl J Med. 2001;344:1058. SOLIRIS™ (eculizumab) [package insert]. Alexion Pharmaceuticals; For Training Purposes Only. Not For Distribution. Alexion Pharmaceuticals, Inc")
87
Extracorpuscular Factors
Antibodies Autoimmune Isoimmune Drugs, antibiotics Fresh water Abnormal plasma lipids Acanthocytosis Venom Snake Spider Bee

88
: - Αμεση (Coombs) test – IgG +3
 test – IgG +3")
89
Αυτοάνοση Αιμολυτική Αναιμία
Μακροκυτταρική Αυξημένα δικτυερυθροκύτταρα Σφαιροκύτταρα Θετική Άμεση Coombs Αυτοάνοση Αιμολυτική Αναιμία

90
Classification Warm Autoimmune (WAIHA) Cold Autoimmune (CAIHA) Mixed
70-80% Cold Autoimmune (CAIHA) 20-30% Mixed 7-8% Paroxysmal Cold Hemoglobinuria rare in adults Drug Induced Hemolytic Anemia
 20-30% Mixed. 7-8% Paroxysmal Cold Hemoglobinuria. rare in adults. Drug Induced Hemolytic Anemia.")
91
Haptenic (e.g. Penicillin, Cephalosporins)
Drug Coats cell; antibody directed against drug/red cell membrane DAT+ for IgG and possibly complement Eluate negative Nonreactive for unexpected antibodies Antibody eluted off red cells reacts with cells+drug but not cells alone Hemolysis develops gradually Discontinue the drug and red cell survival increases

92
Immune Complex (e.g. ceftriaxone)
Acute intravascular hemolysis; renal failure common IgG or IgM antibody Hemolysis due to drug/anti-drug immune complexes that associate with the cell membrane Drug must be present for demonstration of this antibody

93
Drug-independent AIHA (e.g. alpha-methyldopa)
Drug on membrane alters the tertiary structure of the membrane Antibodies are generated against the neoantigen induced by the drug The drug does not need to be present for antibody detection if the membrane has already been altered.

94
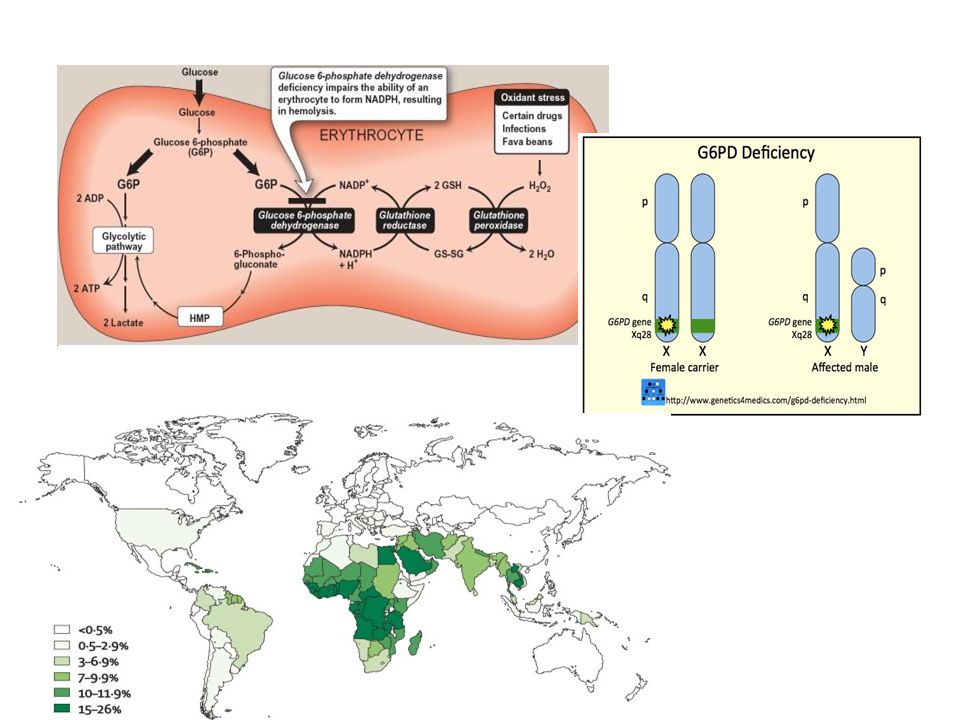
Metabolic Defect G6PD deficiency Pyruvate kinase deficiency
Hexose monophosphate shunt Most common RBC enzyme defect, >50 variants X-linked Low glutathione due to low NADPH Oxidative lysis, Heinz bodies, spherocytic Primaquine, fava beans Pyruvate kinase deficiency Glycolysis Low RBC ATP level Non-spherocytic B12 and folate deficiency Macrocytic HJ bodies Hemoglobinopathies Poikilocytosis Abnormal Hb

95
Schistocytes Malaria

96
Extracorpuscular Factors
Trauma DIC Hemolytic uremic syndrome (HUS) TTP Angiopathy Heat Heart valves “March” hemoglobinuira Microorganisms Malaria Babesia Clostridium Gram negative endotoxin
 TTP. Angiopathy. Heat. Heart valves. March hemoglobinuira. Microorganisms. Malaria. Babesia. Clostridium. Gram negative endotoxin.")
97
Μικροαγγειοπαθητικές αναιμίες

104
The world distribution of haemoglobinopathies overlaps the geographic distribution of malaria. The prevalence has increased in previously non-endemic areas as a consequence of historical and recent immigration flows, slave-trade, trading activities and colonization. In all these regions there is a high prevalence of a thalassaemia. It is believed that carriers of α thalassaemia are protected against malaria and that natural selection is responsible for elevating and maintaining their gene frequencies.

105
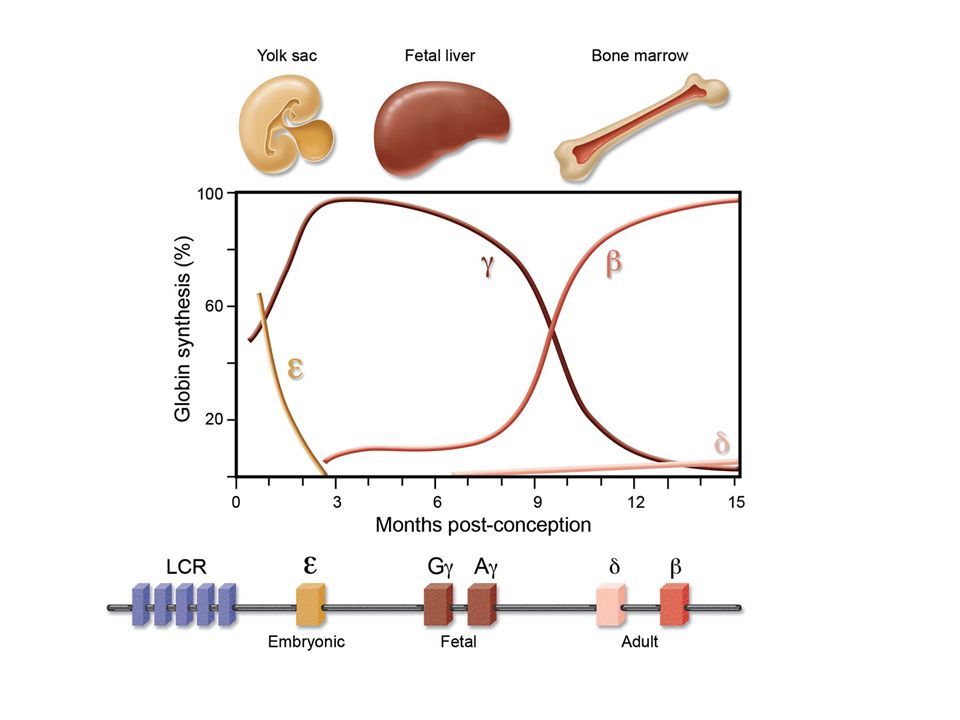
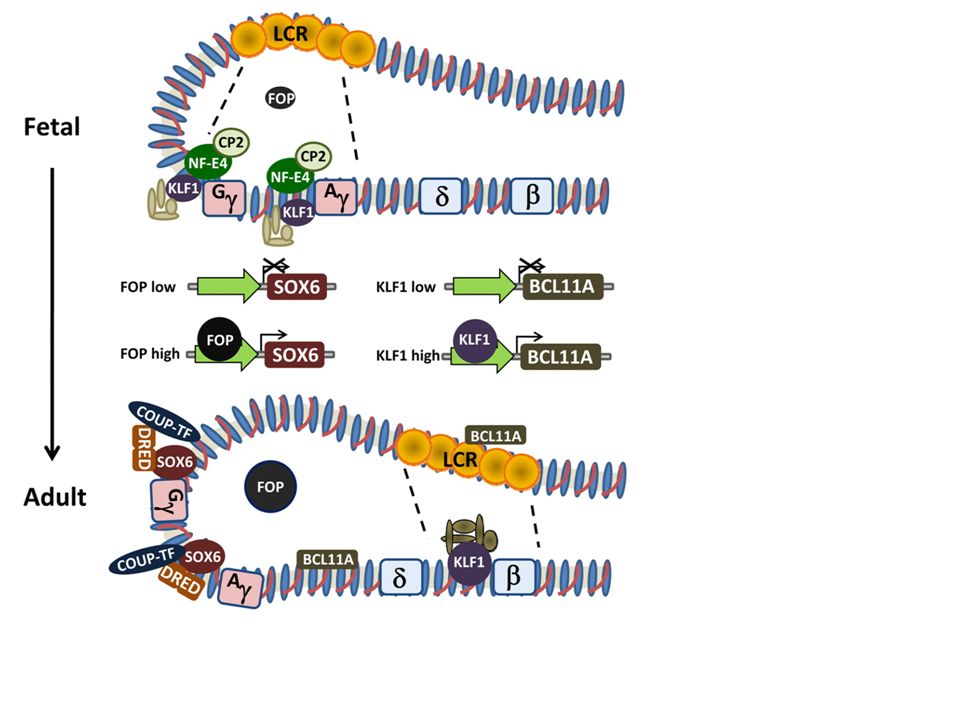
Ανάλογα σε πια, αλυσίδα εκδηλώνεται η γενετική διαταραχή, έχουμε α-Μεσογειακή Αναιμία ή β-Μεσογειακή Αναιμία, καθώς και διάφορους άλλους συνδυασμούς. α-θαλασσαιμία β-θαλασσαιμία

106
The structure of the α-globin gene cluster on chromosome 16
The structure of the α-globin gene cluster on chromosome 16. The telomere is shown as an oval, genes in the region are shown as boxes. The α-globin regulatory region (MCS-R 1 to 4) is indicated as vertical bars. The scale is in kilobases as indicated above. The alpha-gene cluster is enlarged showing the traditional gene names above and the HGVS gene names below. The table below shows the classification of gene defects and phenotypic expression.
 is indicated as vertical bars. The scale is in kilobases as indicated above. The alpha-gene cluster is enlarged showing the traditional gene names above and the HGVS gene names below. The table below shows the classification of gene defects and phenotypic expression.")
107
Τα συνηθέστερα θαλασσαιμικά σύνδρομα
α-θαλασσαιμία-2 (σιωπηλός φορέας) α-θαλασσαιμίες-1 (ετερόζυγη και ομόζυγη) Alfa Thalassemia Minor Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία) Αιμοσφαιρινοπάθεια Bart’s εμβρυϊκός ύδωπας Μείζων α-θαλασσαιμία
 α-θαλασσαιμίες-1 (ετερόζυγη και ομόζυγη) Alfa Thalassemia Minor. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία) Αιμοσφαιρινοπάθεια Bart’s εμβρυϊκός ύδωπας Μείζων α-θαλασσαιμία.")
108
2. α-θαλασσαιμίες-1 (ετερόζυγη και ομόζυγη) Alfa Thalassemia Minor
Εργαστηριακά ευρήματα Υπόχρωμη μικροκυτταρική αναιμία (θαλασσαιμικά ευρήματα) στοχοκυττάρωση ↑ερυθροκυττάρων ↓MCV, ↓MCH HbA2 κφ ή ↓,HbF κφ Ανίχνευση ελεύθερων β-αλυσίδων μετά επώασης και χρώση με (ιώδες του μεθυλίου) 108
 στοχοκυττάρωση. ↑ερυθροκυττάρων. ↓MCV, ↓MCH. HbA2 κφ ή ↓,HbF κφ. Ανίχνευση ελεύθερων β-αλυσίδων μετά επώασης και. χρώση με (ιώδες του μεθυλίου) 108.")
109
2. α-θαλασσαιμίες-1 (ετερόζυγη και ομόζυγη) Alfa Thalassemia Minor
α-θαλαίμασσα-1, πιθανολογείται όταν υπάρχουν β θαλασσαιμικά ευρήματα ενώ τα ευρήματα της ηλεκτροφόρησης είναι φυσιολογικά Διάγνωση γίνεται με μοριακές τεχνικές (DNA ανάλυση)
")
110
Diagram of gap-PCR for detection of α-thalassemia 1 SEA deletion and agarose-gel electrophoresis showing 3 different genotypes from representative blastomeres in the present of relevant controls

111
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία)
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία) Έλλειψη τριών α-γονιδίων (α3) Γονότυπος (α-/--) επίκτητη HbH σε ΟΜΛ (Μ6) » σε μυελοδυσπλαστικά νοσήματα Περίσσεια β-αλυσίδων κατακρήμνιση εντός των ερυθρών φαγοκυττάρωση στο σπλήνα(εξαγγειακή αιμόλυση) διόγκωση του σπλήνα Δημιουργία τετραμερούς β4 (HbH)
 Έλλειψη τριών α-γονιδίων (α3) Γονότυπος (α-/--) επίκτητη HbH σε ΟΜΛ (Μ6) » σε μυελοδυσπλαστικά νοσήματα. Περίσσεια β-αλυσίδων κατακρήμνιση εντός των ερυθρών φαγοκυττάρωση στο σπλήνα(εξαγγειακή αιμόλυση) διόγκωση του σπλήνα. Δημιουργία τετραμερούς β4 (HbH)")
112
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία)
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία) Η HbH (τετραμερές β4) έχει υψηλή δεσμευτική ικανότητα οξυγόνου Δεν παρουσιάζει το φαινόμενο Borh Με αποτέλεσμα ↓↓απόδοση οξυγόνου στους ιστούς
 Η HbH (τετραμερές β4) έχει υψηλή δεσμευτική ικανότητα οξυγόνου. Δεν παρουσιάζει το φαινόμενο Borh. Με αποτέλεσμα ↓↓απόδοση οξυγόνου στους ιστούς.")
113
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία)
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία) Εργαστηριακά ευρήματα-επίχρισμα περιφερικού αίματος Υπόχρωμη μικροκυτταρική αναιμία Στοχοκύτταρα Σχιστοκύτταρα Δακρυοκύτταρα Βασεόφιλη στίξη Ποικιλοκυττάρωση ↑ΔΕΚ, ↑έμμεσης χολερυθρίνης Ευθροκυτταρικά έγκλειστρα (κυανό του κρεζυλίου)
 Εργαστηριακά ευρήματα-επίχρισμα περιφερικού αίματος. Υπόχρωμη μικροκυτταρική αναιμία. Στοχοκύτταρα. Σχιστοκύτταρα. Δακρυοκύτταρα. Βασεόφιλη στίξη. Ποικιλοκυττάρωση. ↑ΔΕΚ, ↑έμμεσης χολερυθρίνης. Ευθροκυτταρικά έγκλειστρα (κυανό του κρεζυλίου)")
114
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία)
3. Αιμοσφαιρινοπάθεια Η (ενδιάμεση α-θαλασσαιμία) Εργαστηριακά ευρήματα Η Ηλεκροφόρηση Αιμοσφαιρίνης και η Υγρή Χρωματογραφία Υψηλής Απόδοσης HPLC(high performance liquid chromatography) HbH 2-40% (συνήθως 8-15%) σημαντικό διαγνωστικό εύρημα HbA2↓ ↓ F↑ (συνήθως 1-3%) HbA (το υπόλοιπο ποσοστό) Hb Bart’s (5%) σε μερικούς ασθενείς
 Εργαστηριακά ευρήματα. Η Ηλεκροφόρηση Αιμοσφαιρίνης και η. Υγρή Χρωματογραφία Υψηλής Απόδοσης HPLC(high performance liquid chromatography) HbH 2-40% (συνήθως 8-15%) σημαντικό διαγνωστικό εύρημα. HbA2↓ ↓ F↑ (συνήθως 1-3%) HbA (το υπόλοιπο ποσοστό) Hb Bart’s (5%) σε μερικούς ασθενείς.")
115
HPLC and Capillary Hb electrophoresis patterns of an adult with HbH disease. The HbH (β4 tetramers) peak elutes from the column as a compressed fraction, and as a fast moving fraction in electrophoresis.
 peak elutes from the column as a compressed fraction, and as a fast moving fraction in electrophoresis..")
116
4. Αιμοσφαιρινοπάθεια Bart’s εμβρυϊκός ύδωπας Μείζων α-θαλασσαιμία
Οφείλεται στην συγκληρονόμηση δύο μεταλλάξεων α0 ΜΑ → ολοκληρωτική απουσία της α-αιμοσφαιρίνης Έλλειψη τεσσάρων α-γονιδίων (α4) Γονότυπος (--/--) Μη συμβατή με τη ζωή, τα έμβρυα πεθαίνουν πριν τη γέννηση τους ή αμέσως μετά (καρδιακή ανεπάρκεια, ατελή οργανογένεση) Σχεδόν όλη η Αιμοσφαιρίνη είναι Hb Bart’s (γ4) Μικρό ποσοστό Αιμοσφαιρίνη Portland (ζ2γ2) Ηλεκτροφόρηση : Hb Bart’s 70-90% Hb H 10-15% Hb Portland 5-10%
 Γονότυπος (--/--) Μη συμβατή με τη ζωή, τα έμβρυα πεθαίνουν πριν τη γέννηση τους ή αμέσως μετά (καρδιακή ανεπάρκεια, ατελή οργανογένεση) Σχεδόν όλη η Αιμοσφαιρίνη είναι Hb Bart’s (γ4) Μικρό ποσοστό Αιμοσφαιρίνη Portland (ζ2γ2) Ηλεκτροφόρηση : Hb Bart’s 70-90% Hb H 10-15% Hb Portland 5-10%")
117
α-θαλασσαιμικά σύνδρομα
ΚΛΙΝΙΚΗ ΕΙΚΟΝΑ ΗΛΕΚΤΡΟΦΟΡΗΤΙΚΑ ΕΥΡΗΜΑΤΑ ΓΟΝΟΤΥΠΟΣ Εμβρυϊκός ύδρωπας Hb Bart (γ4) μη HbH (β4) λειτουργικές Hb Portland α0 /α0 Αιμοσφαιρινοπάθεια Η HbH: 1-30% HbA HbA2 : 1-2% Αιμολυτική αναιμία ποικίλης βαρύτητας α0 /α+ Ετερόζυγη και Ομόζυγη α-ΜΑ HbΑ: κ.φ. MCV, MCH↓ HbA2 : κ.φ.±↓ 3-5% Hb Bart κατά τη γέννηση α0 /α ή α+ /α+ Σιωπηλός φορέας α-ΜΑ HbΑ: κ.φ. MCV, MCH .±↓ ή HbA2 : κ.φ. Οριακά ή κ.φ. α+ /α
 μη. HbH (β4) λειτουργικές. Hb Portland. α0 /α0. Αιμοσφαιρινοπάθεια Η. HbH: 1-30% HbA HbA2 : 1-2% Αιμολυτική αναιμία ποικίλης βαρύτητας. α0 /α+ Ετερόζυγη και Ομόζυγη α-ΜΑ. HbΑ: κ.φ. MCV, MCH↓ HbA2 : κ.φ.±↓ 3-5% Hb Bart κατά τη γέννηση. α0 /α ή. α+ /α+ Σιωπηλός φορέας α-ΜΑ. HbΑ: κ.φ. MCV, MCH .±↓ ή. HbA2 : κ.φ. Οριακά ή κ.φ. α+ /α.")
118
α-Μεσογειακή Αναιμία α-θαλασσαιμία-2 (σιωπηλός φορέας) α-θαλασσαιμία-1
(ελάσσων) Αιμοσφαιρινοπάθεια Η (ενδιάμεση) Ερυθροκυτταρικοί δείκτες MCV MCH Κ.φ./↓ ↓ Επίχρισμα περιφερικού αίματος Κ.φ. Μικροκυττάρωση υποχρωμία Ανισοποικιλοκυττάρωση, υποχρωμία, μικροκυττάρωση, στοχοκύτταρα και σχιστοκύτταρα Ευρήματα από τη γεν.αίματος Σπανιότατα αναιμία Ήπια αναιμία ↑RBC Αναιμία (Hb7-10gr/dl) ΔΕΚ↑ Ερυθροκυττα- ρικά έγκλειστα (β4) ΟΧΙ ΣΥΧΝΑ ΠΑΝΤΑ Ηλεκτροφό- ρηση Hb Κ.φ HbH: 2-40% (συνήθως 8-15%) HbA2:↓ (σπανιότατα Hb Bart’s:5%) Λόγος α/β αλυσίδων
 Αιμοσφαιρινοπάθεια Η. (ενδιάμεση) Ερυθροκυτταρικοί δείκτες MCV MCH. Κ.φ./↓ ↓ Επίχρισμα. περιφερικού. αίματος. Κ.φ. Μικροκυττάρωση. υποχρωμία. Ανισοποικιλοκυττάρωση, υποχρωμία, μικροκυττάρωση, στοχοκύτταρα και σχιστοκύτταρα. Ευρήματα από. τη γεν.αίματος. Σπανιότατα. αναιμία. Ήπια αναιμία. ↑RBC. Αναιμία (Hb7-10gr/dl) ΔΕΚ↑ Ερυθροκυττα- ρικά έγκλειστα (β4) ΟΧΙ. ΣΥΧΝΑ. ΠΑΝΤΑ. Ηλεκτροφό- ρηση Hb. Κ.φ. HbH: 2-40% (συνήθως 8-15%) HbA2:↓ (σπανιότατα Hb Bart’s:5%) Λόγος α/β. αλυσίδων.")
119
β-ΘΑΛΑΣΣΑΙΜΙΕΣ

120
Οι μεταλλάξεις οι οποίες περιορίζουν τη σύνθεση των β αλυσίδων αιμοσφαιρίνης είναι γνωστές ως μεταλλάξεις β+ (βήτα συν) ΜΑ, ενώ αυτές οι οποίες καταργούν ολοκληρωτικά τη σύνθεσή τους ως μεταλλάξεις β0 (βήτα μηδέν) ΜΑ. Κάποιες μεταλλάξεις που επιτρέπουν σε ένα μεγάλο βαθμό τη σύνθεση των β αλυσίδων είναι γνωστές ως μεταλλάξεις β++.
 ΜΑ. Κάποιες μεταλλάξεις που επιτρέπουν σε ένα μεγάλο βαθμό τη σύνθεση των β αλυσίδων είναι γνωστές ως μεταλλάξεις β++.")
121
Οι περισσότερες μεταλλάξεις β0 και β+ έχουν
επίπτωση στον αιματολογικό φαινότυπο των φορέων όπως: ↑ RBC ↓ (MCV 60-70fl) ↓(MCH 19-23pg), ↑(Hb A2) συνήθως μεταξύ 4-6%.
 ↓(MCH 19-23pg), ↑(Hb A2) συνήθως μεταξύ 4-6%.")
124
ΟΜΟΖΥΓΗ β-ΘΑΛΑΣΣΑΙΜΙΑ Ή ΜΕΙΖΟΝ β-ΘΑΛΑΣΣΑΙΜΙΑ THALASEEMIA MAJOR Ή ΝΟΣΟΣ ΤΟΥ COOLEY
Πρόκειται για ασθενείς με ομζυγωτία (β0β0, β+β+) ή συνδυασμένη ετεροζυγωτία (β0β+) β θαλασσαιμίας Η ζωή των ασθενών εξαρτάται άμεσα από τη μετάγγιση αίματος ήδη από την πρώιμη παιδική ηλικία
 ή συνδυασμένη ετεροζυγωτία (β0β+) β θαλασσαιμίας Η ζωή των ασθενών εξαρτάται άμεσα από τη μετάγγιση αίματος ήδη από την πρώιμη παιδική ηλικία")
125
Παθοφυσιολογία των κλινικών εκδηλώσεων

126
ΟΜΟΖΥΓΗ β-ΜΕΣΟΓΕΙΑΚΗ ΑΝΑΙΜΙΑ

128
ΜΕΤΑΓΓΙΣΕΙΣ

129
θεραπεία Μεταγγίσεις αίματος Αποσιδήρωση Συμπτωματική Αγωγή
Σπληνεκτομή (ενδεχομένως) Μεταμόσχευση του μυελού των οστών Γονιδιακή θεραπεία (διαφαίνεται πραγματοποιήσιμη στο μέλλον)
 Μεταμόσχευση του μυελού των οστών. Γονιδιακή θεραπεία (διαφαίνεται πραγματοποιήσιμη στο μέλλον)")
130
Από τι πεθαίνουν οι ασθενείς με β-ΜΑ?

131
Tissue Iron Concentrations in Transfusion-dependent Thalassemia Patients
Thyroid Fe = 1.6 – 6.8% d wt Heart Fe = 0.6 –1.3% d wt Liver Fe = % d wt Pancreas Fe = % d wt Adapted from Modell & Berdoukas, 1984 131

132
Ενδιάμεση β-θαλασσαιμία (thalassemia intermedia)
Γονότυπος (β+/β+ ή β0/β++) Ενδιάμεση κλινική κατάσταση μεταξύ ετερόζυγης –μείζονος ΜΑ Μέτρια Αναιμία Δεν απαιτείται μετάγγιση αίματος Μακρά επιβίωση
 Ενδιάμεση κλινική κατάσταση μεταξύ ετερόζυγης –μείζονος ΜΑ. Μέτρια Αναιμία. Δεν απαιτείται μετάγγιση αίματος. Μακρά επιβίωση.")
133
Ενδιάμεση β-θαλασσαιμία (thalassemia intermedia) διαφορική διάγνωση
Μείζων β θαλασσαιμία Ενδιάμεση β θαλασσαιμία Κλινική Εικόνα Ηλίκα εμφάνισης <2 ετών >2ετών Επίπεδα Hb 2-7 g/dl 7-10 g/dl Διόγκωση σπληνός/ήπατος έντονη Μέτρια έως έντονη Εργαστηριακή εικόνα Hb F >50% 10-50% (μπορεί και 100%) Hb A2 <4% >4%
 Hb A2. <4% >4%")
134
Δρεπανοκυτταρικά Σύνδρομα

135
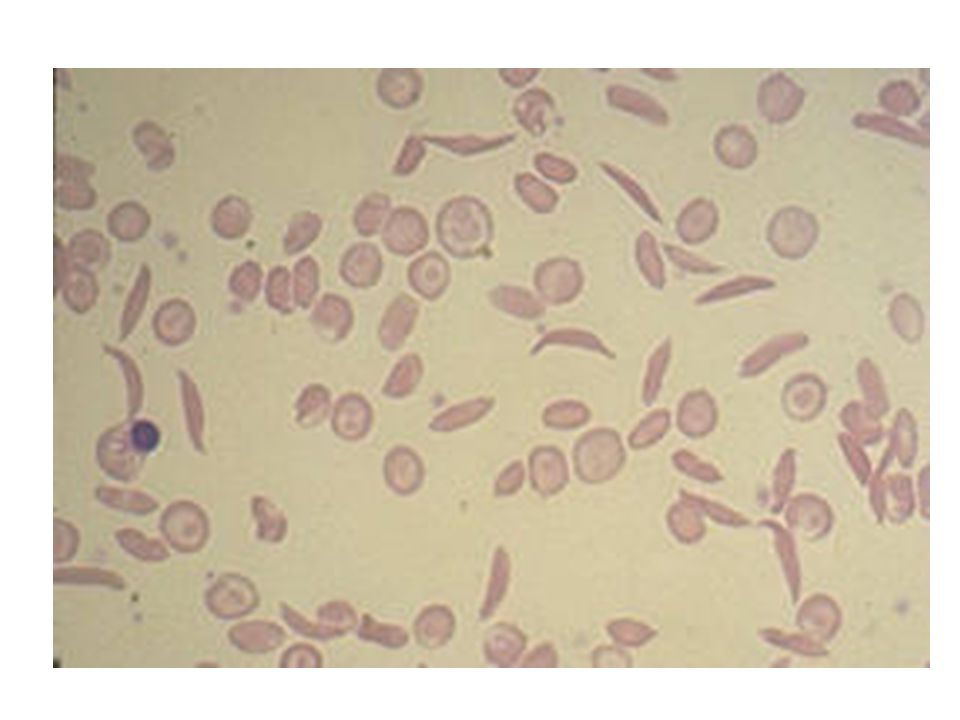
Η δρεπανοκυτταρική αναιμία δημιουργείται από μια μετάλλαξη του γονιδίου που κωδικοποιεί τη β-πολυπεπτιδική αλυσίδα της αιμοσφαιρίνης, α. Στο μοντέλο του μορίου της αιμοσφαιρίνης φαίνεται η θέση της μετάλλαξης, β. φυσιολογικό ερυθροκύτταρο, επάνω και δρεπανοειδές ερυθροκύτταρο, κάτω.

136
Δρεπανοκυτταρικά Σύνδρομα
Παθολογική φυσιολογία Τα δρεπανοκύτταρα εμποδίζουν τη φυσιολογική κυκλοφορία του αίματος στα τριχοειδή αγγεία δημιουργώντας προβλήματα σε διάφορα όργανα όπως στο σπλήνα και τους πνεύμονες. Τα δρεπανοκύτταρα καταστρέφονται ταχύτερα με φαγοκυττάρωση από τα μακροφάγα του σπλήνα (εξαγγειακή αιμόλυση) με συνέπεια την εμφάνιση συμπτωμάτων αναιμίας.
 με συνέπεια την εμφάνιση συμπτωμάτων αναιμίας.")
137
Τα δρεπανοκύτταρα λόγω του σχήματος δεν μπορούν εύκολα να περάσουν από τη μικροκυκλοφορία διαφόρων οργάνων , σχηματίζουν μικρά αθροίσματα και προκαλούν έμφρακτα στα όργανα αυτά .

138
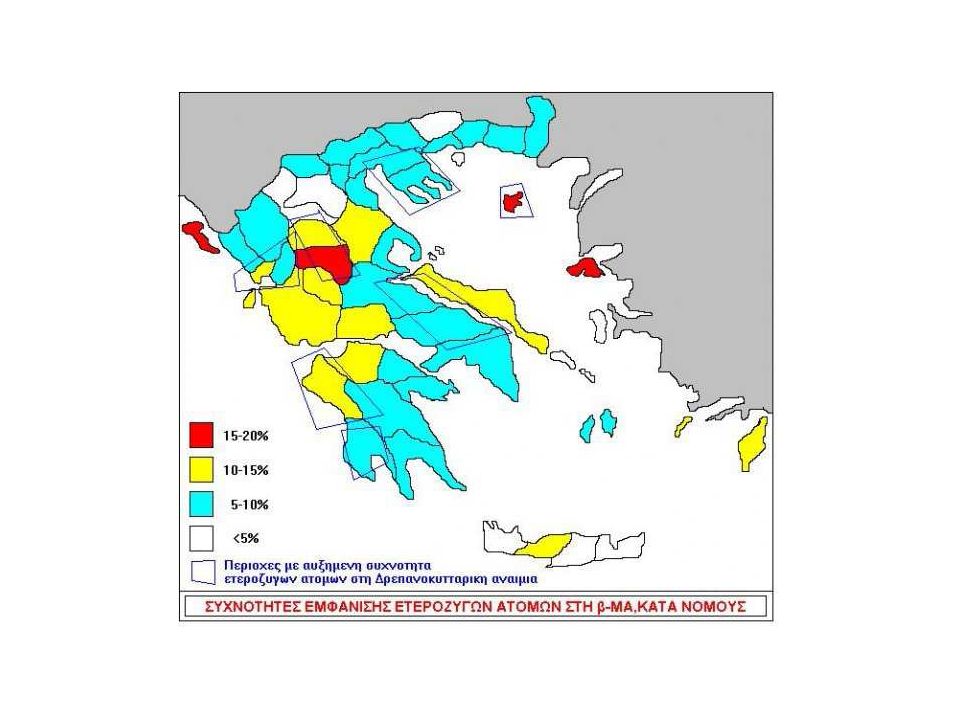
Δρεπανοκυτταρικά Σύνδρομα Γεωγραφική κατανομή

139
Sickle Cell Anemia and Malaria
Children with sickle trait (heterozygotes) have a milder course of P. falciparum. However, children with SS disease have more severe courses with a very high mortality rate. 139
 have a milder course of P. falciparum. However, children with SS disease have more severe courses with a very high mortality rate")
140
Ετερόζυγη Δρεπανοκυτταρική Αναιμία
Γονότυπος (β/βs) Συχνότητα στην Ελλάδα 1% Άτομα ασυμπτωματικά Σε συνθήκες υποξίας : υψηλό υψόμετρο 3,000m αεροπορικά ταξίδια μικροέφρακτα στο έντονη άσκηση μυελό των νεφρών αιματουρία υψηλός πυρετός
 Συχνότητα στην Ελλάδα 1% Άτομα ασυμπτωματικά. Σε συνθήκες υποξίας : υψηλό υψόμετρο 3,000m. αεροπορικά ταξίδια μικροέφρακτα στο. έντονη άσκηση μυελό των νεφρών αιματουρία. υψηλός πυρετός.")
141
Ετερόζυγη Δρεπανοκυτταρική Αναιμία εργαστηριακά ευρήματα
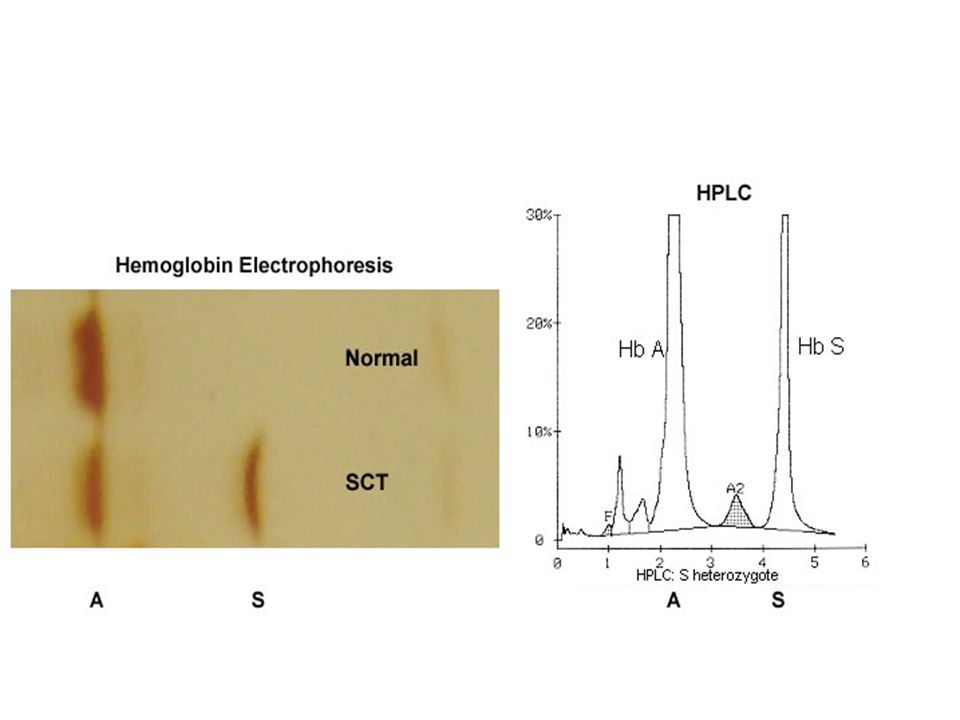
MCV, MCH, Hb ΚΦ/↓(μικρή ελάττωση) Επίχρισμα περιφερικού αίματος ΚΦ Μικροκύτταρα, Στοχοκύτταρα(σπάνια) Ηλεκτροφόρηση Hb HbS: 35-45%, HbA:50-60%, HbA2≤3,HbF<2%(δεν επηρεάζονται) Δοκιμασία δρεπάνωσης Θετική Γενική Ούρων Αιματουρία (ελαφρά) Υποσθενουρία
 Επίχρισμα περιφερικού αίματος. ΚΦ. Μικροκύτταρα, Στοχοκύτταρα(σπάνια) Ηλεκτροφόρηση Hb. HbS: 35-45%, HbA:50-60%, HbA2≤3,HbF<2%(δεν επηρεάζονται) Δοκιμασία δρεπάνωσης. Θετική. Γενική Ούρων. Αιματουρία (ελαφρά) Υποσθενουρία.")
142
Ομόζυγη Δρεπανοκυτταρική αναιμία (Sickle cell anemia)
Οι ομοζυγώτες φέρουν δύο παθολογικά γονίδια Γονότυπος βs/βs Χαρακτηρίζεται από Hb S: 70-95%, HbA2<3, Hb A=0

143
Οξεία Απλαστική Κρίση . Κρίση εγκλωβισμού

144
Αγγειοαποφρακτική ή Επώδυνη Κρίση
Επώδυνες κρίσεις συνήθως, στην κοιλιά στο θώρακα και τα οστά , που οφείλονται σε Αγγειοαποφρακτικά φαινόμενα (σε συνθήκες ανοξίας) (1η σε συχνότητα αιτία θανάτου) Αίτια : Ψύχος ↑ Ζέστη Αφυδάτωση Λοιμώξεις (30%) κακουχία κ.ά. Εργαστηριακά ευρήματα ↓ Hb ↑ΔΕΚ ↑MCHC ↑RDW ↑ Εμπύρηνων Ερυθροκυττάρων ↑ WBC ↑ PLT
 (1η σε συχνότητα αιτία θανάτου) Αίτια : Ψύχος. ↑ Ζέστη. Αφυδάτωση. Λοιμώξεις (30%) κακουχία κ.ά. Εργαστηριακά ευρήματα. ↓ Hb. ↑ΔΕΚ. ↑MCHC. ↑RDW. ↑ Εμπύρηνων Ερυθροκυττάρων. ↑ WBC. ↑ PLT.")
145
Θεραπευτική Αντιμετώπιση
Θεραπευτική Αντιμετώπιση Ο ασθενής πρέπει να αισθάνεται άνετα Χορηγείται αποτελεσματική Αναλγησία Χορηγείται Οξυγόνο εφόσον υπάρχει Υποξία (SaO2 <92%) Εξασφάλιση επαρκής Ενυδάτωσης Χορηγείται Αντιβίωση εφόσον υπάρχει λοίμωξη Χορηγείται Αίμα εφόσον υπάρχει ένδειξη Αντιμετωπίζονται τα ειδικά Κλινικά Προβλήματα Το προσδόκιμο όριο ζωής αγγίζει τα 50 έτη
 Εξασφάλιση επαρκής Ενυδάτωσης. Χορηγείται Αντιβίωση εφόσον υπάρχει λοίμωξη. Χορηγείται Αίμα εφόσον υπάρχει ένδειξη. Αντιμετωπίζονται τα ειδικά Κλινικά Προβλήματα. Το προσδόκιμο όριο ζωής αγγίζει τα 50 έτη.")
146
Definitive Treatment: Hydroxyurea therapy
Indicated for children >5y/o who have severe complications of SCD Effective because increases HbF, decreases leukocytes, platelets and reticulocytes CBC must be monitored regularly when on therapy for leukopenia 146

147
ΜΙΚΡΟΔΡΕΠΑΝΟΚΥΤΤΑΡΙΚΗ ΑΝΑΙΜΙΑ
Η Μικροδρεπανοκυτταρική Αναιμία είναι ο συνδυασμός ετερόζυγης β-θαλασσαιμίας και δρεπανοκυτταρικής αναιμίας Γονότυπος βs/βo Κλινική εικόνα : Αναιμία, Υπίκτερο , Σπληνομεγαλία, Επώδυνες Αιμολυτικές Κρίσεις Εργαστηριακά ευρήματα: Μορφολογία ερυθρών τύπου β-θαλασσαιμίας Ηλεκτροφόρηση Hb: HbS>85% ↑ HbA2, ↑HbF, HbA=0

148
ΜΙΚΡΟΔΡΕΠΑΝΟΚΥΤΤΑΡΙΚΗ ΑΝΑΙΜΙΑ
Γονότυπος βs/β+ Μέτρια Αναιμία Επώδυνες Αιμολυτικές Κρίσεις (σπάνια) Ηλεκτροφόρηση Hb: HbS=55-75% HbA=10-30% HbA2>3.5% HbF5-30%
 Ηλεκτροφόρηση Hb: HbS=55-75% HbA=10-30% HbA2>3.5% HbF5-30%")
149
Pathophysiology of Polycythemia

150
Secondary Polycythemia
Appropriate EPO (tissue/kidney hypoxia) pulmonary disease high altitude congenital heart disease abnormal hemoglobin high affinity carboxyhemoglobin
 pulmonary disease. high altitude. congenital heart disease. abnormal hemoglobin. high affinity. carboxyhemoglobin.")
151
Secondary Polycythemia
Inappropriate EPO (ectopic production) Tumors (hepatoma, renal carcinoma, leiomyoma, hamartoma) Renal disorders (transplantation, cysts) hemangiomas Androgen abuse EPO abuse Familial polycythemia
 Tumors (hepatoma, renal carcinoma, leiomyoma, hamartoma) Renal disorders (transplantation, cysts) hemangiomas. Androgen abuse. EPO abuse. Familial polycythemia.")
152
Oxygen delivery vs. Hematocrit
J Clin Invest 1963;42:1150

153
P. Vera - Symptoms & Signs
Headache Weakness Pruritis (aquagenic) Dizziness Diaphoresis Visual disturbance Weight loss Signs Splenomegaly 70% Skin plethora 67% Hepatomegaly 40% Conjunctival plethora 59% Systolic Hypertension 72%
 Dizziness. Diaphoresis. Visual disturbance. Weight loss. Signs. Splenomegaly 70% Skin plethora 67% Hepatomegaly 40% Conjunctival plethora 59% Systolic Hypertension 72%")
154
P. Vera - Diagnosis (PVSG criteria)
RBC mass elevated SaO2 > 92% Splenomegaly (or) thrombocytosis Leukocytosis high LAP high B12 Significance True vs. spurious R/O most 2 causes Evidence for MPD False Positive 0.5% smokers, drinkers
 thrombocytosis. Leukocytosis. high LAP. high B12. Significance. True vs. spurious. R/O most 2 causes. Evidence for MPD. False Positive 0.5% smokers, drinkers.")
155
P. vera - Bone Marrow Biopsy

156
P. Vera - Natural History

159
Calreticulin

Παρόμοιες παρουσιάσεις


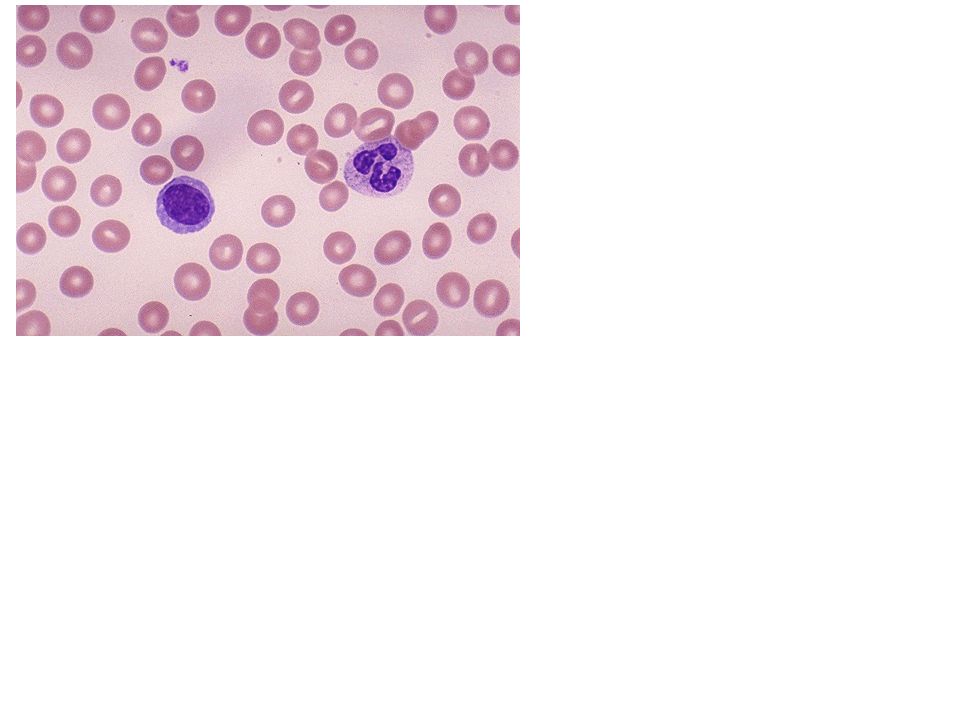
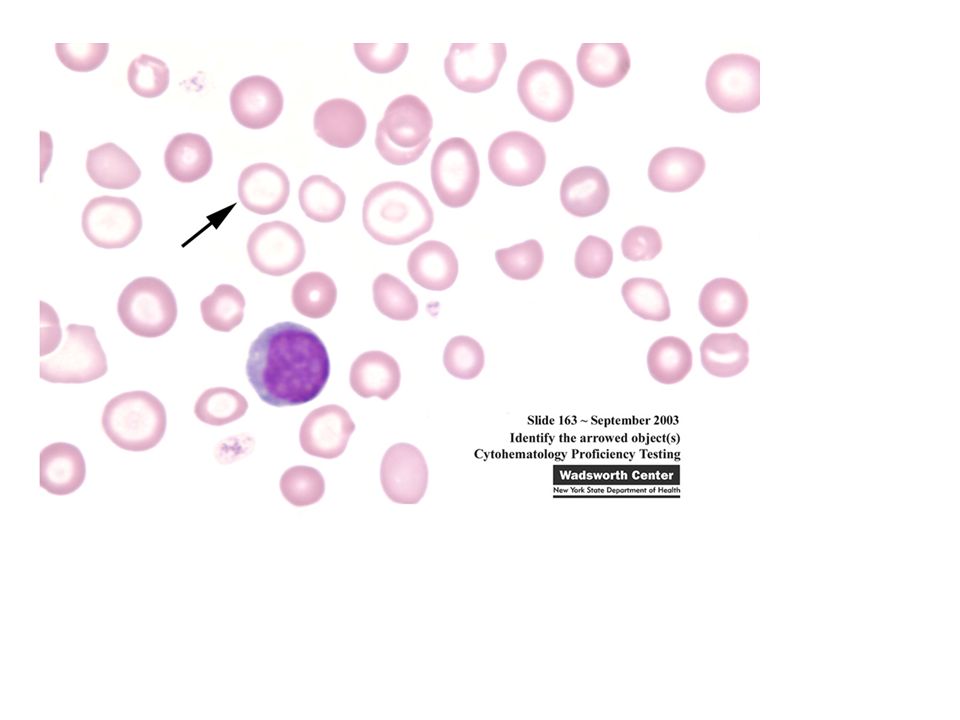
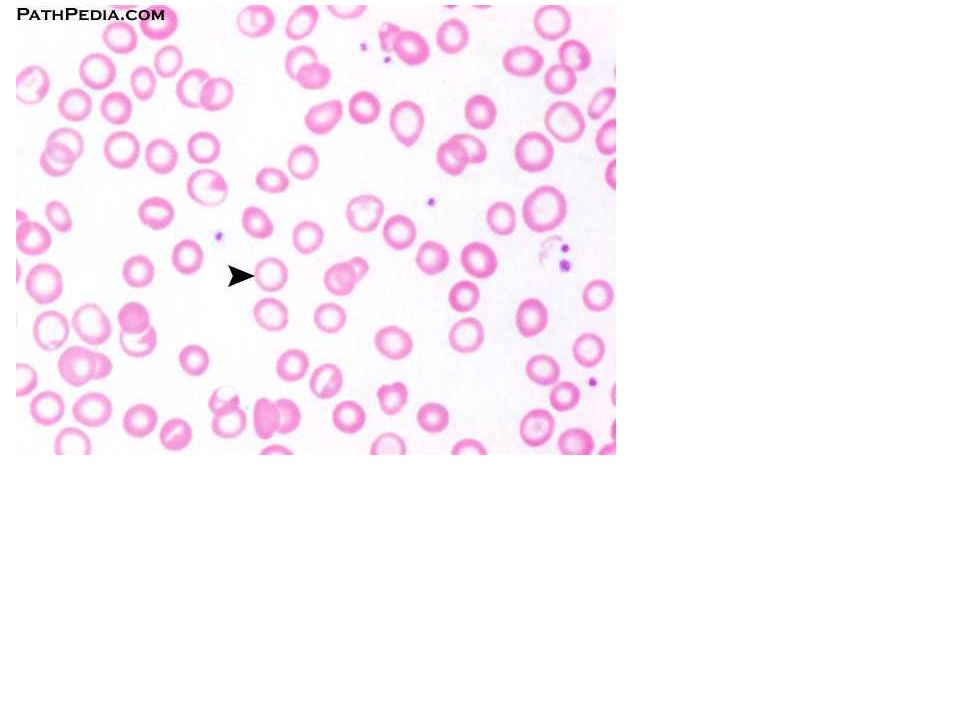
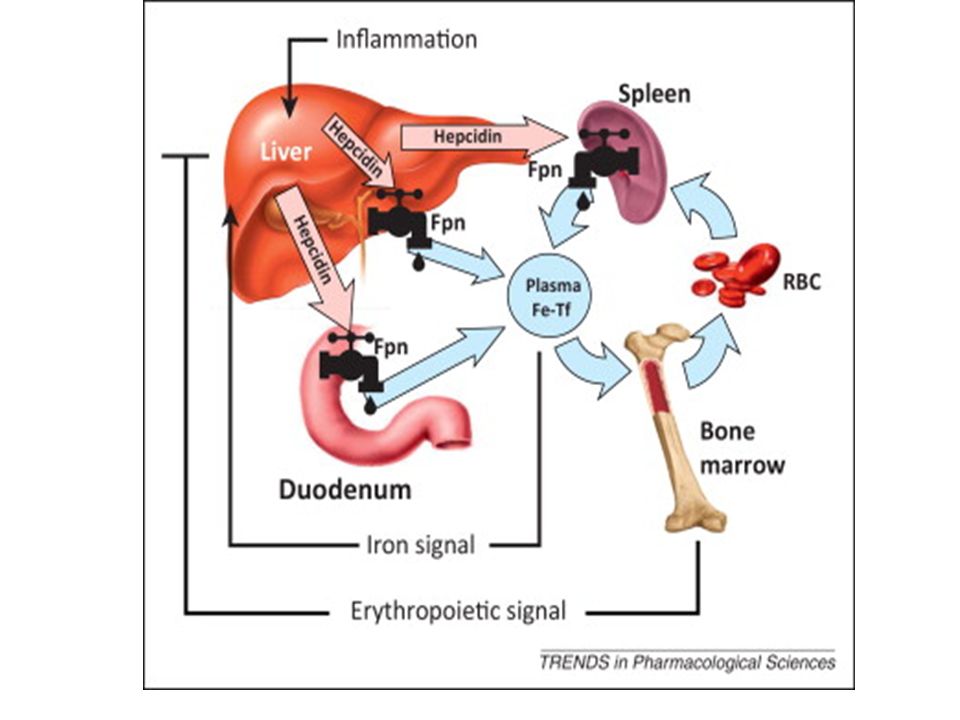


















 Ενότητα 5: Αναιμία Χρόνιας Νόσου Αναστάσιος Κριεμπάρδης Τμήμα Ιατρικών εργαστηρίων Ανοικτά Ακαδημαϊκά Μαθήματα στο ΤΕΙ Αθήνας Το περιεχόμενο.>")


 Αριθμός δικτυοερυθροκυττάρων Αμεση και έμμεση Coombs.>")



